

Primaire laterale sclerose (PLS): een consensus over diagnostische criteria
15-02-2020
Ingekorte versie
Samenvatting:
Primaire laterale sclerose (PLS) is een neurodegeneratieve aandoening van het volwassen motorisch systeem. De diagnose wordt gekenmerkt door een traag progressief syndroom van de bovenste motorneuronen, en is klinisch, na uitsluiting van structurele, neurodegeneratieve en metabolische imitatievormen. Het onderscheid tussen PLS en vormen van amyotrofische laterale sclerose met predominantie in de bovenste motorneuronen blijft een aanzienlijke uitdaging in de vroege symptomatische fase van beide aandoeningen, waarbij nog steeds getwist wordt over de vraag of ze al dan niet een klinisch en histopathologisch continuüm vormen. De huidige diagnostische criteria voor PLS kunnen een barrière vormen voor therapeutische ontwikkeling, want er liggen lange periodes tussen de aanvang van de symptomen en de formele diagnose. Hoewel het mogelijk is dat nieuwe technologieën die gevoelig zijn aan de betrokkenheid van zowel de bovenste als de onderste motorneuronen uiteindelijk de controverse rond de diagnose van PLS kunnen opklaren, presenteren we hier de bijgewerkte consensus rond diagnostische criteria, die als doel hebben de diagnostische vertraging te reduceren, het ontwerp van klinische studies te optimaliseren en de ontwikkeling van ziektewijzigende therapieën te katalyseren.
Het wezenlijke klinische syndroom:
Er is sprake van consistente klinische observaties die worden gerapporteerd aan de hand van meerdere gevalsreeksen van PLS. De gemiddelde leeftijd bij de aanvang van de symptomen bedraagt ongeveer 50 jaar. Dat is ten minste tien jaar vroeger dan non-familiale ALS, en tien jaar later dan HSP (hereditaire spastische parapese). Er zijn gevallen bekend waarbij de symptomen hun aanvang nemen in de kindertijd, maar vele daarvan kunnen we tegenwoordig koppelen aan monogenetische of ontwikkelingsaandoeningen. Bij PLS is stelselmatig een mannelijke predominantie vastgesteld (reikwijdte 2–4:1). PLS is verraderlijk in zijn aanvang, zodat individuen meestal pas laat na het intreden van de vroegste symptomen terugvallen op gespecialiseerde diensten. Bij de meeste patiënten doen de eerste symptomen zich voor in de onderste ledematen. Bij een significante minderheid gaat het echter om de corticobulbaire trajecten, gepaard gaande met dysarthrie en vaak prominente emotionaliteit (pseudobulbair affect). Hoewel dysfagie op de voorgrond kan treden, is de waarde van gastrostomie veel onduidelijker dan bij ALS, en is de behoefte aan non-invasieve ventilatie bij PLS eerder uitzonderlijk. De betrokkenheid van de onderste ledematen in de vroege symptomatische fase kan zich voordoen in de vorm van een gevoel van onevenwicht of een stroevere gang. Daarbij moet niet noodzakelijk sprake zijn van een prominente sensorische betrokkenheid. Bij onderzoeken stelt men steeds weer spasticiteit met pathologische hyperreflexie vast. Hoewel PLS normaal gezien na verloop van tijd ook de bovenste ledematen aantast, blijft een focale symptomatische aanvang in de bovenste ledematen erg uitzonderlijk bij deze ziekte.
Kader 1: een consensus van diagnostische criteria voor primaire laterale sclerose (PLS)1. Hoofdprincipes Een diagnose met PLS vereist: A. de aanwezigheid van: B. de afwezigheid van: 2. diagnostische zekerheid • ► Waarschijnlijke PLS wordt gedefinieerd door de afwezigheid van significante actieve OMN degeneratie 2–4 jaar sinds de aanvang van de symptomen. *Klinische tekenen, waaronder spasticiteit en de daaraan verbonden zwakte, pathologische hyperreflexie (waaronder het teken van Hoffman en bilaterale-extensor-teenresponsen), pseudobulbair affect. Labobewijsmateriaal van BMN-dysfunctie van recente neuro-imaging-, neurofysiologische en neurochemische biomerkers (zie het hoofdstuk ‘Opkomende technologie’) moet nog worden gevalideerd. †Minimaal verhoogde insertieactiviteit en positieve scherpe golven of gevallen van fibrillatiepotentieel in spieren van de uitersten zijn toegestaan (zie het hoofdstuk ‘Elektromyografische overwegingen’). |
Kader 2 Genetische varianten die werden gerapporteerd bij een klein percentage van syndromen met predominantie in de bovenste motorneuronenSPG7* Primaire laterale sclerose blijkt in wezen een sporadische aandoening te zijn, waarbij de diagnose voornamelijk wordt gebaseerd op klinische kenmerken, eerder dan op genotype. |
Conclusies:
Ontwikkelingen op het vlak van neuro-imaging, neurofysiologie en moleculaire biologie hebben geen afbreuk gedaan aan de status die PLS van oudsher heeft als een op zichzelf staand, klinisch gedefinieerd syndroom. De zeldzaamheid en het langdurige overlevingstraject ervan hebben in vergelijking met ALS geresulteerd in een belangrijke mate van verwaarlozing op het vlak van klinische studies. De traditionele toevlucht tot EMG met overinterpretatie van minimale merkers van OMN-betrokkenheid draagt mogelijk bij tot significante diagnostische vertraging. De ontwikkeling van een internationaal register dat iedereen met ‘waarschijnlijke PLS’ omvat, zal een preciezere afbakening mogelijk maken van de pathogenese ten opzichte van ALS, en therapeutische ontwikkelingen in een stroomversnelling brengen. Afgezien van primaire ziektewijzigende therapie, is de meest dringende onbevredigde behoefte voor patiënten met PLS wellicht de behandeling van kernsymptomen, eerder dan een extensie van de overlevingsduur. De ontwikkeling van meer effectieve vormen van leniging van spasticiteit, zonder dat de spierkracht daarbij in het gedrang komt, zou een grote impact hebben op mensen die leven met PLS, ongeacht de nood op de lange termijn aan neuroprotectieve of regeneratieve therapie. Vanuit de erkenning dat vooruitgang op het gebied van moleculaire fenotypering mogelijk belangrijker wordt dan een zuiver klinische diagnose, hopen we dat deze pragmatische criteria een groter vertrouwen zullen kweken, zodat de diagnostische vertraging kan worden weggewerkt. Zodoende kunnen patiënten al gebruik maken van potentieel ziektewijzigende therapieën in een vroeger stadium van hun onvermogen.
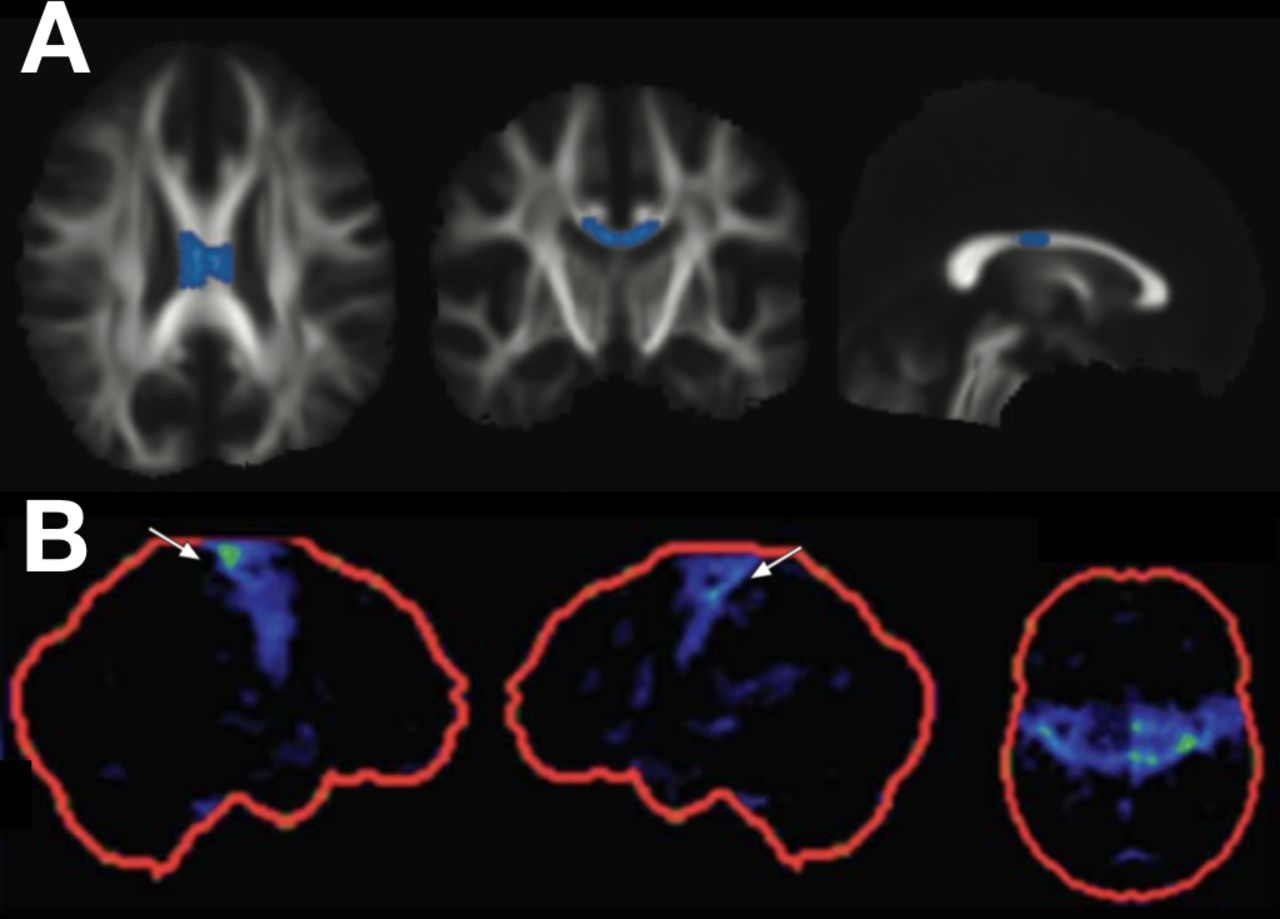
Figuur 1
Bevindingen inzake diffusietensor-imaging en fluorodeoxyglucose (FDG) positron-emissietomografie (PET) in gevallen van primaire laterale sclerose (PLS). De gemiddelde diffusiviteit in het middelste gedeelte van het corpus callosum is verhoogd in gevallen van PLS vergeleken met amyotrofische laterale sclerose (rij A, PLS- vs. ALS-groepbevindingen, geaccentueerd op axiale, coronale en sagittale beelden van het skelet van de wittestofwegen; met dank aan de auteur, zie Iwata et al). Focaal hypometabolisme kan worden vastgesteld in de primaire motorische cortices bij PLS (rij B, FDG PET z-scorebeelden in de rechter sagittale, linker sagittale en axiale vlakken; met dank aan de auteur, zie Claassen et al). Vooralsnog heeft geen van deze technieken voldoende sensitiviteit of specificiteit om ze op zichzelf toe te passen voor de diagnose van PLS.
Vertaling: Bart De Becker
Bron: BMJ - Journal of Neurology, Neurosurgery, and Psychiatry