

Recente vorderingen op het gebied van de pathogenese van en therapieën voor amyotrofische laterale sclerose
11-04-2021
Amyotrofische laterale sclerose (ALS) is een progressieve en zeer ontwrichtende neurodegeneratieve aandoening, met een incidentie van 1 tot 5 gevallen per 100.000 inwoners. De ziekte wordt gekenmerkt door spierdenervatie, zwakte, atrofie en verlamming, wat binnen de 3 tot 5 jaar na de diagnose vaak leidt tot de dood van de patiënt (door ademhalingsfalen). ALS vangt aan met een progressieve afname van het aantal bovenste en onderste motorneuronen, wat wordt aangezwengeld door een complexe etiologie (genen en omgevingsfactoren). Sporadische vormen van de ziekte komen het meest voor (tot 90% van de gevallen), maar ze vallen klinisch en histopathologisch niet te onderscheiden van de verschillende familiale vormen die tot dusver werden beschreven en waarbij mutaties in meer dan 25 ALS-gerelateerde genen zijn betrokken. Om verscheidene redenen zijn de meest relevante daarvan: (i) SOD1, dat de cruciale antioxidant enzym superoxide dismutase‐1 encodeert; (ii) TARDBP en FUS, die respectievelijk de eiwitten TAR‐DNA bindend proteïne‐43 (TDP‐43) of gefuseerd in sarcoom (FUS) encoderen en die zijn betrokken bij de splitsing, het transport en de stabiliteit van pre‐mRNA; en (iii) C9orf72, dat een eiwit encodeert dat betrokken is bij de intracellulaire uitwisseling van neuronen en andere celfuncties die we nog niet helemaal doorgronden. Veranderingen in SOD‐1, TARDBP, FUS en C9orf72 doen zich voor in de meeste gevallen (zowat 70%) van familiale ALS (Kim, Gautier, Tassoni‐Tsuchida, Ma, & Gitler, 2020).
SOD1 was in 1993 het eerste ALS‐gerelateerde gen dat werd geïdentificeerd (Rosen et al., 1993), en de ontdekking ervan was een belangrijke stimulans voor het onderzoek naar ALS. Dit was deels te danken aan de snelle ontwikkeling van transgene dierenmodellen (de meeste ervan bij muizen) die overexpressie vertoonden van de verschillende menselijke SOD1-mutaties (het meest voorkomende was G93A; Ripps, Huntley, Hof, Morrison, & Gordon, 1995). Deze deden jarenlang dienst als het enige instrument voor de studie van de pathogenese van en therapieën voor ALS. De aard van deze stimulans blijkt duidelijk uit de gegevens die zijn verwerkt in Figuur 1, die de ontwikkeling toont van het aantal artikels die werden gepubliceerd omtrent ALS-studies en die gedurende de afgelopen 50 jaar werden verzameld in PubMed. Zoals blijkt uit Figuur 1 zorgden de daaropvolgende ontdekkingen van nieuwe, met ALS gelinkte genen (TARDBP, FUS, en C9orf72) voor een verdere groei van het aantal ALS-studies, ook in dit geval door transgene modellen te leveren met een overexpressie van de meest frequente mutaties die worden teruggevonden in deze genen (Van Damme, Robberecht, & Van Den Bosch, 2017).
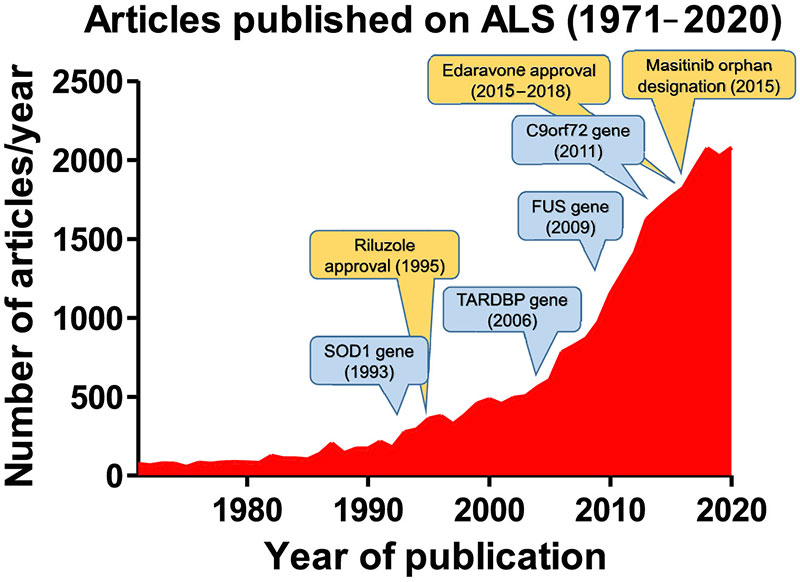
FIGUUR 1
Vordering van de publicatie van het aantal ALS gerelateerde artikels gedurende de afgelopen 50 jaar, op basis van een zoekopdracht in PubMed aan de hand van de term ‘amyotrophic lateral sclerosis’, met vermelding van het publicatiejaar van de ontdekking van de meest relevante genen die zijn gelinkt met ALS en de goedkeuring van de schaarse geneesmiddelen die ALS-patiënten ter beschikking hebben.
Zoals hierboven aangestipt, zijn sporadische en familiale ALS-gevallen niet van elkaar te onderscheiden en hetzelfde geldt voor de pathogene mechanismen die ten grondslag liggen aan motorneuronendood. De progressieve ontdekking van de verschillende ALS gerelateerde genen en de ontwikkeling van experimentele modellen die zijn gebaseerd op deze genen hebben bijgedragen tot een beter begrip van deze pathogene mechanismen, zoals blijkt uit Figuur 2.
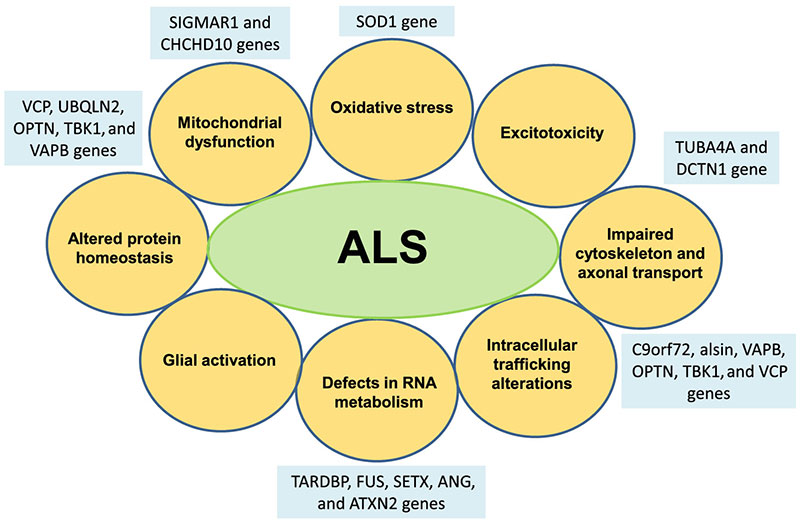
FIGUUR 2
Diagram van de verschillende pathogene mechanismen die zijn betrokken bij de degeneratie van motorneuronen in geval van ALS en hun verband met enkele van de verschillende genen die gelinkt zijn met ALS.
ALS is een ziekte met een duidelijke onbevredigde therapeutische behoefte aan effectieve geneesmiddelen, aangezien diegene die momenteel voorhanden zijn slechts in beperkte mate effectief zijn: ze verlengen de levensduur slechts voor enkele maanden. Tot deze therapeutische middelen behoren het anti‐excitotoxische riluzol (Rilutek®), dat in 1995 werd goedgekeurd en gedurende lange tijd het enige beschikbare ALS-geneesmiddel was. In 2015 werd het antioxiderende middel edaravone (Radicava®) goedgekeurd in Japan en vervolgens in de Verenigde Staten (2017) en Canada (2018). Nog in 2015 werd de anti‐inflammatoire tyrosinekinase-remmer masitinib (Kinavet‐CA1®) ontworpen als weesgeneesmiddel voor de behandeling van ALS. Dit zijn de enige ziekte wijzigende geneesmiddelen die patiënten tot hun beschikking hebben. Gedurende jaren werden ook veel andere potentiële therapeutische middelen, met verschillende actiemechanismen (anti‐excitotoxisch, antioxiderend, anti‐inflammatoir, op de mitochondria gericht, neurotrofe factoren en andere middelen) klinisch onderzocht. En hoewel deze middelen in dierenmodellen een goed profiel vertoonden inzake veiligheid en efficiëntie, hielden deze voordelen jammer genoeg niet stand in klinisch onderzoek (zie Figuur 3, een samenvatting van enkele van de meest relevante therapeutische middelen die de afgelopen jaren op klinisch niveau werden geëvalueerd). Een brede waaier van factoren, waaronder de voorspelbaarheid van de experimentele modellen, het late stadium waarin deze klinische studies werden gestart, de heterogeniteit van de pathogene mechanismen die zich voordoen bij ALS of de behoefte aan strategieën met meerdere doelwitten, worden in overweging genomen als mogelijke redenen voor deze mislukkingen.

FIGUUR 3
Voorbeelden van families van samenstellingen die werden onderzocht op hun potentiële neuroprotectieve (ziekte wijzigende) eigenschappen in geval van ALS en van families van samenstellingen die het klinische onderzoekstadium bereikten met een ander resultaat (adaptatie naar Fernández‐Ruiz, 2019)
De identificatie gedurende de afgelopen 15 jaar van nieuwe ALS gerelateerde genen heeft cruciale informatie opgeleverd over de moleculaire en cellulaire mechanismen die de pathologie veroorzaken of bijdragen aan het ziekteverloop. Deze ontdekkingen hebben bijgedragen aan de identificatie van nieuwe doelwitten voor de ontwikkeling van neuroprotectieve therapieën. Bovendien hebben deze nieuwe genen ALS gekoppeld aan andere neurodegeneratieve pathologieën, zoals frontotemporale degeneratie (FTD), zodat beide ziekten momenteel worden beschouwd als twee zijden van dezelfde pathologische medaille. Het doel van dit themanummer is al deze recente vorderingen op het gebied van ALS en aanverwante pathologieën samen te brengen en zo de wetenschappelijke gemeenschap een bijgewerkt overzicht aan te reiken van de etiologie van ALS, de pathogene mechanismen die zijn betrokken bij de degeneratie van motorneuronen en de huidige ontwikkeling van nieuwe en efficiënte therapieën.
In dit themanummer, dat voortvloeit uit de ‘International Workshop on ALS: new genes, new treatments, new hopes’ (dat plaatsvond in Madrid op 30 en 31 oktober 2019, met de steun van het BJP) vindt de lezer om te beginnen een overzichtsartikel van Javier Riancho, Adolfo López de Munaín en medewerkers over een nieuwe conceptualisering van de etiologie van ALS. De auteurs beschrijven ALS als een complexe ziekte die wordt veroorzaakt door individueel genetisch risico, het verouderingsproces en de invloed van omgevingsfactoren, aspecten die interageren om de drempelwaarde te bepalen van de aanvang, het verloop en de prognose van de ziekte. Ze bespreken de relevantie voor de ziekte van de omgevingsvoorwaarden die zouden kunnen worden gewijzigd en overlopen het bewijsmateriaal voor kanker, auto-immuniteit en metabole aandoeningen als zijnde belangrijke factoren die leiden tot ALS (Riancho et al., 2020).
In een tweede overzichtsartikel behandelen Riancho en medewerkers ALS vanuit het gezichtspunt van een ziekte die het motorische systeem overstijgt, met de nadruk op het sensorische systeem. Ze combineren informatie afkomstig van patiënten en van dierenmodellen en onderstrepen de schade aan sensorisch‐motorische netwerken als een belangrijke factor die bij deze ziekte maar al te vaak over het hoofd werd gezien of ondergewaardeerd (Riancho, Paz‐Fajardo, & López de Munaín, 2020).
In het daaropvolgende artikel verzamelen Rosario Osta en medewerkers het experimentele bewijsmateriaal dat werd gegenereerd in hun en andere laboratoria. Dit materiaal schraagt de opvatting dat schade aan perifere weefsels – met de focus op de skeletspier – een belangrijke factor is. Ze beoordelen de rol die bij deze ziekte wordt gespeeld door schade aan de mitochondriale functie van spieren, het energiemetabolisme, proteostase en het RNA-metabolisme, alsook de rol die wordt gespeeld door tekortkomingen in de myogenese. De meeste informatie die door de auteurs werd verzameld in dit overzicht werd verkregen uit muterend-SOD‐1-muizen, het klassieke ALS-model, dat gebaseerd is op het eerste gen dat werd geïdentificeerd in verband met ALS. Ze behandelen ook de inspanningen die worden geïnvesteerd in de ontwikkeling van nieuwe therapeutische strategieën die zich richten op de skeletspier om de aanvang en het verloop van deze ziekte op te houden (Manzano et al., 2020).
Het ribonucleo-eiwit TDP‐43, dat in 2006 werd ontdekt, wordt erg vaak teruggevonden bij ALS (en ook bij FTD), in de vorm van aggregaten en de cytosol, en dit niet alleen bij familiale gevallen die te wijten zijn aan mutaties van het TARDBP-gen, maar ook bij veel sporadische gevallen (tot 95% van alle ALS-gevallen). Dit is het gevolg van een waaier van posttranslationele wijzigingen (bv. alomtegenwoordigheid, fosforylering, acetylering, sumoylering en splitsing) die zich voordoen in TDP‐43, wat leidt tot de conformationele wijziging ervan, tot disregulering, een veranderde cellulaire locatie, gehinderde functie, afzetting en aggregatie. Deze fenomenen werden uitgebreid bestudeerd door Emanuele Buratti in zijn bijdrage aan dit themanummer. Daarin benadrukt hij het therapeutisch potentieel van verschillende lage MW-samenstellingen, die in staat zijn de pathologische kenmerken van TDP‐43 te wijzigen, waaronder de expressieniveaus, cytoplasmische verkeerde lokalisering, posttranslationele wijzigingen, splitsing, stressgranulenrekrutering en andere kenmerken (Buratti, 2020).
In dezelfde richting van het overzicht van Buratti gaat het overzicht van Ana Martinez en medewerkers, dat zich concentreert op de remming van verscheidene kinasen bij de fosforylering van TDP‐43 en andere belangrijke ALS gerelateerde eiwitten als potentiële ALS-therapie. Daarbij ligt de nadruk op c-kit (een tyrosinekinase die wordt geremd door masitinib, dat momenteel een weesgeneesmiddelentoewijzing heeft als ALS-behandeling), ROCK (bv. fadusil), mTOR (bv. rapamycine), GSK-3 (bv. lithium), en andere remmers die al deel uitmaken van het klinische scenario. Het overzichtsartikel behandelt ook vele andere stoffen die een mate van efficiëntie vertoonden en redelijk veilig bleken in preklinische modellen (bv. remmers voor CK‐1, CDC7 en MAP3K) en zelfs in vroege stadia van de ontdekking van geneesmiddelen, zoals remmers voor de Tau-tubulinekinasen (TTBK’s) en de van cycline afhankelijke kinasen (CDK’s) (Palomo, Nozal, Rojas‐Prats, Gil, & Martinez, 2020).
Sigma‐1-receptoren worden geëncodeerd door het SIGMAR1-gen, dat eveneens wordt gelinkt aan de ontwikkeling van ALS. Als deel van hun verschillende celsubstraten in het centrale zenuwstelsel, komen sigma‐1-receptoren vooral voor in motorneuronen en in intracellulaire complexen die worden gevormd door het endoplasmische reticulum en de mitochondria. Ze doen dienst als begeleidende eiwitten en moduleren essentiële processen voor de overleving van motorneuronen. Deze receptoren als doelwit kiezen kan dan ook nuttig zijn om het afsterven van deze neuronen tegen te gaan. Dit aspect wordt uitgebreid behandeld in het overzichtsartikel van Xavier Navarro en medewerkers, dat de neuroprotectieve eigenschappen wil valideren van verscheidene liganden voor deze receptoren (Herrando‐Grabulosa, Gaja‐Capdevila, Vela, & Navarro, 2020).
ALS wordt ook gelinkt met epigenetische basiskenmerken. Het therapeutische potentieel van dit verband is dan ook recent geëxploreerd aan de hand van verscheidene onderzoekslijnen. Ludo Van den Bosch en medewerkers bieden in dit themanummer een overzicht van het potentieel van een ingreep op histone deacetylases, een welbepaalde klasse van epigenetische enzymen. Ze exploreren het potentieel van gekende remmers zoals valproate, lithium en resveratrol, alsmede een aantal nieuwe synthetische remmers, voor de verschillende subtypes van histone deacetylases, waaronder de sirtuïnes, en dit in preklinische modellen en een paar klinische studies, waarbij ze bijzondere aandacht besteden aan hun vermogen om de hersen-bloedbarrière te overschrijden en aan hun verdraagbaarheid en veiligheid (Klingl, Pakravan, & Van Den Bosch, 2020).
We mogen hopen dat deze bijdragen leiden tot het inschakelen van nog meer hulpbronnen en tot nog meer inspanningen die vereist zijn om verder te gaan met de identificatie van nieuwe therapeutische doelwitten en nieuwe therapeutische stoffen, zodat de voortdurende zoektocht naar geneesmiddelen die klinisch effectief zijn bij het vertragen van het ziekteverloop van ALS in stand wordt gehouden. De patiënten hebben dringend nood aan deze inspanningen.
Vertaling: Bart De Becker
Bron: British Journal of Pharmacology