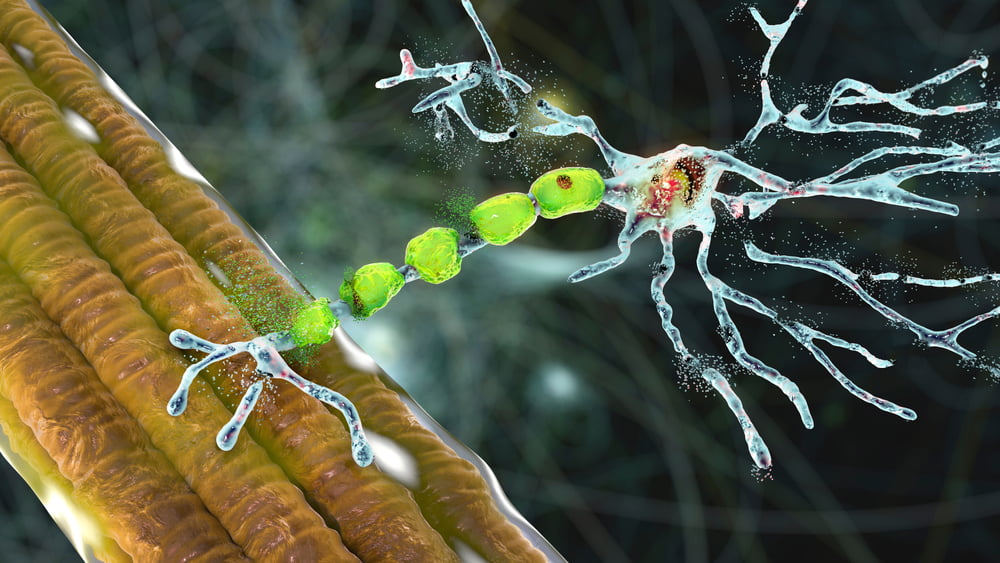
Amyotrophe Lateralsklerose (ALS) ist eine tödliche Nerven-Muskel-Erkrankung, die alle Muskelgruppen mit Ausnahme der autonom gesteuerten Muskeln befallen kann. Ein ALS-Patient stirbt durchschnittlich 33 Monate nach der Diagnose durch Lähmung der Atem- oder Schluckmuskulatur. Es gibt jedoch eine erhebliche Streuung in der endgültigen Lebenserwartung von ALS-Patienten.
| ALS | Sklerose der äußeren Nervenbahnen im Rückenmark zusammen mit Muskelatrophie |
| Atrofie | Absterben durch Mangel an Reizen |
| Myo- | Muskel |
| Amyotrofie | Muskelverlust |
| Lateraal | Lateral seitlich, von den Seitensträngen |
| Sclerose | Verhärtung |






ALS ist eine nicht ansteckende Erkrankung, deren Ursache noch nicht mit Sicherheit festgestellt wurde. Bis heute ist keine adäquate Behandlungs- oder Präventionsmethode bekannt. Die durchschnittliche Überlebenszeit nach der Diagnose und der spezifische Krankheitsverlauf unterscheiden sich von Patient zu Patient, was die Prognose erschwert. Daher ist es wichtig, dass Patienten sich nicht blind auf Durchschnittswerte verlassen. Warum sollten sie nicht die Ausnahme sein? Realistisches Denken ist natürlich wünschenswert, aber man sollte auch nicht in Schwarzmalerei verfallen. Leider sind Stress und Müdigkeit Faktoren, die dazu führen können, dass sich die Krankheit schneller entwickeln können.
Im Jahr 1862 verwendete Jean-Marie Charcot erstmals den Begriff „Amyotrophe Lateralsklerose“, um eine Beeinträchtigung sowohl des ersten als auch des zweiten Motoneurons anzuzeigen. Im Jahr 1874 gab er die erste genaue Beobachtung des Krankheitsbildes ALS. In einigen Ländern wird ALS auch MND genannt: Motor (Bewegung), Neuron (Nervenzelle), Disease (Krankheit).
In den Vereinigten Staaten ist die Erkrankung auch als „Lou-Gehrig-Krankheit“ bekannt, nach dem Namen eines legendären Baseballspielers, der 1941 an dieser Krankheit starb.