Was bedeuten ALS, PLS und PSMA?
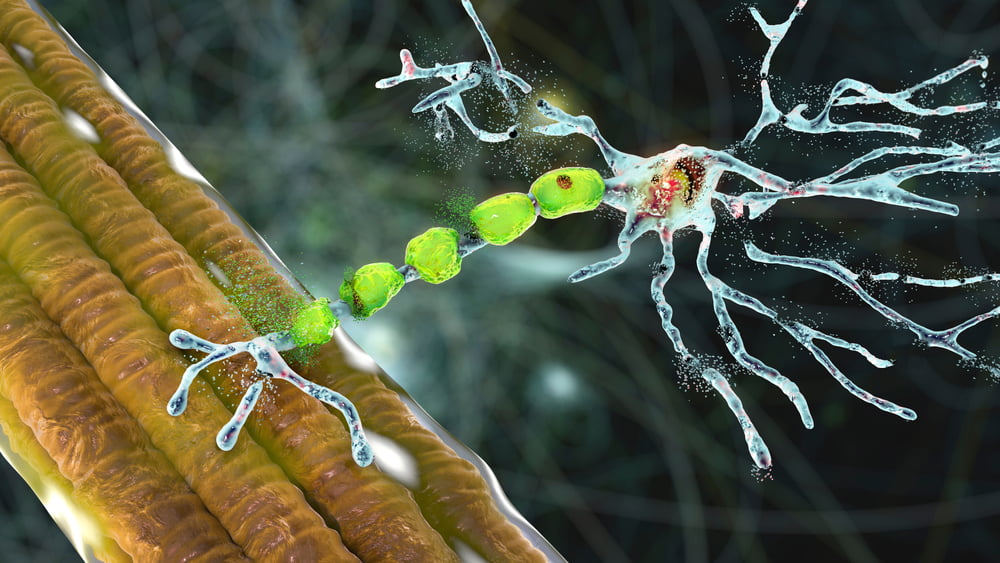
Die Planung einer Bewegung findet in einer Reihe von komplizierten Netzwerken im Gehirn statt, deren Funktionsweise unter anderem bei Krankheiten wie Parkinson und Huntington gestört ist. Um eine Bewegung auszuführen und damit Muskelkraft zu erzeugen, musst du zwei Arten von Nervenzellen aktivieren. Die erste Art von Nervenzellen befindet sich im Gehirn (in der grauen motorischen Substanz, die als Kortex oder Rinde bezeichnet wird); die zweite Art befindet sich im Rückenmark, um die Muskeln der Arme, Beine und des Rumpfes zu benutzen, oder im Hirnstamm (dem unteren Teil des Gehirns), um die Muskeln des Schluckens und der Sprache zu benutzen. Da diese Nervenzellen (Neuronen genannt) für die motorische Aktivität verantwortlich sind, werden sie als Motoneuronen bezeichnet. Neuronen im Gehirn werden als höhere oder zentrale Motoneuronen bezeichnet, weil sie sich im zentralen Nervensystem befinden. Neuronen im Hirnstamm oder Rückenmark werden als untere oder periphere motorische Neuronen bezeichnet.

Abbildung 1: Obere motorische Neuronen im Gehirn und untere motorische Neuronen im Hirnstamm und Rückenmark .
die unteren Neuronen des Hirnstamms und des Rückenmarks
Die oberen Motoneuronen senden elektrische Signale über Kabel an die unteren Motoneuronen. Diese Kabel sind lange Äste des Neurons, die Axone genannt werden. Zusammen bilden diese Axone die kortikospinale Bahn, die von der Hirnrinde bis zum Rückenmark (lateinisch „medulla spinalis“) verläuft. Die elektrischen Signale erreichen und aktivieren die unteren Motoneuronen. Diese wiederum senden ihre Äste oder Axone zu den Muskeln. Diese Axone bilden die Nerven, die vom Rückenmark zu den Muskeln verlaufen. Diese Axone sind manchmal sehr lang. Während das Neuron selbst nur einen Zehntel Millimeter im Durchmesser hat, kann ein Axon leicht einen Meter lang sein (also 10.000 Mal länger). Schließlich muss es zum Beispiel vom Rückenmark zu den Fußmuskeln gelangen (Abbildung 2).
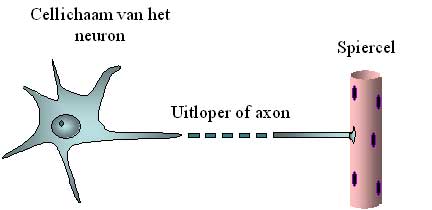
Abbildung 2: Schematische Darstellung des Zellkörpers des Motoneurons und seines Axons.
Das Axon ist leicht 10.000-mal länger als der Durchmesser des Zellkörpers, weshalb es unterbrochen dargestellt ist.
Bei der körperlichen Untersuchung stellt der Neurologe fest, ob die motorischen Neuronen normal funktionieren. Das EMG (kurz für Elektromyografie) kann Anomalien in den peripheren Neuronen aufzeigen, die bei der körperlichen Untersuchung gar nicht erkennbar sind.
Diese beiden Neuronen funktionieren nicht mehr richtig und verkümmern („degenerieren“) bei einer Motoneuronenkrankheit oder Motorneuronendegeneration. Die bekannteste dieser Krankheiten ist ALS (Amyotrophe Lateralsklerose), aber es gibt mehrere Arten. Eine Fehlfunktion der oberen Motoneuronen führt vor allem zu Spastizität und abnormal scharfen Reflexen; ein Ausfall der unteren Motoneuronen führt zu Muskelschwäche und -schwund. Der letztgenannte Begriff bezieht sich auf die Ausdünnung eines Muskels, die z. B. an der Hand deutlich sichtbar ist.
Bei ALS werden beide Arten von Neuronen (zentrale und periphere) krank und sterben ab. Das Ergebnis ist eine Kombination von Symptomen: Muskelschwund aufgrund der Degeneration der peripheren Neuronen und gleichzeitig abnorme Reflexe und Spastizität, die auf die Degeneration der zentralen Neuronen hinweisen. Diese Kombination und die kontinuierliche Verschlimmerung der Krankheit ermöglichen es dem Arzt, ALS zu diagnostizieren. Das EMG wird bestätigen, dass die peripheren Neuronen betroffen sind.
In den Medien werden ALS und andere Motoneuronenerkrankungen oft als „Muskelkrankheiten“ bezeichnet. Das ist verständlich, denn Muskelfunktionsstörungen sind natürlich am sichtbarsten. Aber es sind nicht die Muskeln, die „schief“ gehen, sondern die Nervenzellen, die sie mit den nötigen Signalen versorgen müssen.
Bei einigen Patienten beschränkt sich die Krankheit zunächst auf die unteren Nervenzellen. Diese Menschen entwickeln Muskelschwund und Kraftverlust, aber keine Spastik. Bei einigen dieser Patienten entwickeln sich dann recht schnell die Anzeichen einer Erkrankung der oberen Motoneuronen: Sie haben tatsächlich ALS. Ein anderer Teil der Patienten entwickelt jedoch nie Anzeichen einer zentralen Motoneuronenerkrankung. Neurologen haben keinen passenden Namen für diese Krankheit. Sie wird untere Motoneuronenerkrankung, progressive spinomuskuläre Atrophie (PSMA) oder progressive Muskelatrophie (PMA) genannt. Diese Krankheit schreitet tendenziell langsamer voran als ALS, aber das ist nicht wirklich bekannt. Es ist durchaus möglich, dass es sich bei dieser Form nur um eine Variante der ALS handelt: Schließlich entwickeln manche Menschen mit einer Mutation im SOD1-Gen nie eine Spastik und haben trotzdem ALS.
Ein kleiner Teil der Patienten entwickelt nur Anomalien der unteren Motoneuronen in den Armen und Anomalien der oberen Motoneuronen in den unteren Gliedmaßen. Dies wird als „Flail-Arm-Syndrom“ bezeichnet. Wir wissen, dass diese Krankheit recht langsam fortschreitet. Erst in einem sehr späten Stadium sind die Sprach- und Schluckmuskeln betroffen.
Auch das Gegenteil ist der Fall. Bei manchen Patienten sind nur die zentralen Motoneuronen betroffen. Diese Form der Motoneuronenerkrankung wird als primäre Lateralsklerose (PLS) bezeichnet. Nach einer gewissen Zeit wird jedoch bei einigen dieser Patienten deutlich, dass auch die unteren Motoneuronen betroffen sind: Diese Menschen haben dann ALS. Um die Entwicklung von ALS zu erkennen, unterziehen sich ALS-Patienten regelmäßig EMG-Tests.
Der andere Teil der Patientenpopulation entwickelt nie ALS. Sie haben wirklich ALS. Wir wissen nicht viel über diese Krankheit. Wir wissen, dass sie immer noch zehnmal seltener ist als ALS. Es wird geschätzt, dass es in Flandern nur etwa fünfzig ALS-Patienten gibt. Es ist jedoch wichtig, die Krankheit richtig zu diagnostizieren und sie von ALS zu unterscheiden. Die Aussichten für PLS-Patienten sind anders: Die Krankheit schreitet viel langsamer voran als ALS. Eine Krankheitsdauer von zehn oder sogar fünfzehn Jahren ist bei PDS nicht ungewöhnlich, bei ALS hingegen schon.