
Que signifient ALS, PLS, PSMA ?
La planification d’ un mouvement se produit dans une série de réseaux compliqués dans le cerveau, dont le fonctionnement se dérègle dans des maladies telles que Parkinson et Huntington, entre autres. Pour effectuer un mouvement et donc générer de la puissance musculaire, tu dois activer deux types de cellules nerveuses. Un premier type de cellules nerveuses se trouve dans le cerveau (dans la matière grise motrice appelée cortex ou écorce) ; un deuxième type dans la moelle épinière pour utiliser les muscles des bras, des jambes et du tronc, ou dans le tronc cérébral (la partie inférieure du cerveau) pour utiliser les muscles de la déglutition et de la parole. Comme ces cellules nerveuses (appelées neurones) sont responsables de l’activité motrice, elles sont appelées neurones moteurs. Les neurones du cerveau sont appelés neurones moteurs supérieurs ou centraux car ils sont situés dans le système nerveux central. Les neurones du tronc cérébral ou de la moelle épinière sont appelés neurones moteurs inférieurs ou périphériques.

Figure 1 : Les motoneurones supérieurs du cerveau et les motoneurones inférieurs du tronc cérébral et de la moelle épinière .
les neurones inférieurs du tronc cérébral et de la moelle épinière
Les motoneurones supérieurs envoient des signaux électriques par des câbles aux motoneurones inférieurs. Ces câbles sont de longues ramifications du neurone, appelées axones. L’ensemble de ces axones constitue la voie corticospinale en termes médicaux, car elle va du cortex à la moelle épinière, appelée « medulla spinalis » en latin. Les signaux électriques atteignent et activent les neurones moteurs inférieurs. Ces derniers envoient à leur tour leurs ramifications ou axones vers les muscles. Ces axones forment les nerfs qui vont de la moelle épinière aux muscles. Ces axones sont parfois très longs. Alors que le neurone lui-même ne mesure qu’un dixième de millimètre de diamètre, un axone peut facilement mesurer un mètre de long (c’est-à-dire 10 000 fois plus). Après tout, il doit aller de la moelle épinière aux muscles du pied, par exemple (figure 2).
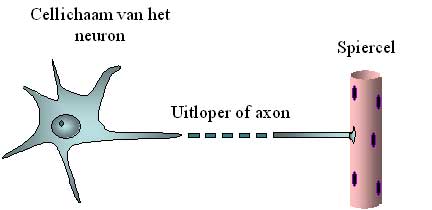
Figure 2: Représentation schématique du corps cellulaire du motoneurone et de son axone.
L’axone est facilement 10 000 fois plus long que le diamètre du corps cellulaire, c’est pourquoi il est représenté interrompu.
Lors de l’examen physique, le neurologue détermine si les neurones moteurs fonctionnent normalement. L’EMG (abréviation d’électromyographie) peut identifier des anomalies des neurones périphériques qui ne sont même pas détectables à l’examen physique.
Ce sont ces deux neurones qui fonctionnent mal et qui languissent plus tard (« dégénèrent ») dans les troubles des neurones moteurs ou dégénérescences des neurones moteurs. Parmi ces types de maladies, la SLA (sclérose latérale amyotrophique) est la plus connue, mais il en existe plusieurs types. Le mauvais fonctionnement des motoneurones supérieurs donne principalement lieu à de la spasticité et à des réflexes anormalement vifs ; la défaillance des motoneurones inférieurs donne une faiblesse musculaire et une atrophie musculaire. Ce dernier terme signifie l’amincissement d’un muscle, bien visible par exemple au niveau de la main.
Dans la SLA, les deux types de neurones (centraux et périphériques) deviennent malades et dépérissent. Il en résulte une combinaison de symptômes : fonte des muscles due à la dégénérescence des neurones périphériques, et en même temps réflexes anormaux et spasticité indiquant la dégénérescence des neurones centraux. Cette combinaison ainsi que l’aggravation continue de la maladie permettent au médecin de diagnostiquer la SLA. L’EMG confirmera l’atteinte des neurones périphériques.
Dans les médias, la SLA et les autres maladies du motoneurone sont souvent décrites comme des « maladies musculaires ». C’est compréhensible, car le dysfonctionnement des muscles est naturellement le plus visible. Cependant, ce ne sont pas les muscles qui « vont mal », mais les cellules nerveuses qui doivent leur fournir les signaux nécessaires.
Chez certains patients, la maladie se limite d’abord aux neurones inférieurs. Ces personnes développent une atrophie musculaire et une perte de force, mais pas de spasticité. Chez certains de ces patients, les signes de la maladie du motoneurone supérieur se développent ensuite assez rapidement : ils sont réellement atteints de la SLA. Cependant, une autre proportion de patients ne développe jamais de signes de maladie du motoneurone central. Les neurologues n’ont pas de nom approprié pour désigner cette maladie. Elle est appelée maladie du motoneurone inférieur, ou atrophie spinomusculaire progressive (PSMA) ou atrophie musculaire progressive (PMA). Cette maladie a tendance à évoluer plus lentement que la SLA, mais cela n’est pas vraiment bien connu. Il est tout à fait possible que cette forme ne soit en fait qu’une variante de la SLA : après tout, certaines personnes présentant une mutation du gène SOD1 ne développent jamais de spasticité et sont tout de même atteintes de la SLA.
Une petite proportion de patients ne développe que des anomalies des motoneurones inférieurs dans les bras et des anomalies des motoneurones supérieurs dans les membres inférieurs. C’est ce qu’on appelle le « syndrome du bras ballant ». On sait que cette maladie évolue assez lentement. Ce n’est que très tard dans son évolution que les muscles de la parole et de la déglutition sont touchés.
L’inverse existe également. Chez certains patients, seuls les motoneurones centraux sont touchés. Cette forme de maladie du motoneurone est appelée sclérose latérale primaire (SLP). Cependant, au bout d’un certain temps, il apparaît clairement chez certains de ces patients que les motoneurones inférieurs sont également touchés : ces personnes sont alors atteintes de SLA. Pour détecter l’évolution vers la SLA, les patients atteints de SLA subissent régulièrement des EMG.
L’autre partie des patients ne développe jamais cette évolution vers la SLA. Ils sont vraiment atteints de la SLA. On ne sait pas grand-chose sur cette maladie. Nous savons qu’elle est encore 10 fois moins fréquente que la SLA. On estime qu’il n’y a qu’une cinquantaine de patients atteints de SLA en Flandre. Cependant, il est important de la diagnostiquer correctement et de la distinguer de la SLA. En effet, les perspectives pour les patients atteints de SDP sont différentes : la maladie évolue beaucoup plus lentement que la SLA. Une durée de la maladie de dix, voire quinze ans, n’est pas inhabituelle pour la SDP, mais elle l’est pour la SLA.