
What do ALS, PLS and PSMA mean?
Planning a movement takes place in a series of complicated networks in the brain, the functioning of which is disrupted in diseases such as Parkinson’s and Huntington’s, among others. To perform a movement and therefore generate muscle power, you need to activate two types of nerve cells. The first type of nerve cell is found in the brain (in the grey motor matter known as the cortex or bark); the second type is found in the spinal cord to use the muscles of the arms, legs and trunk, or in the brain stem (the lower part of the brain) to use the muscles of swallowing and speech. As these nerve cells (called neurons) are responsible for motor activity, they are called motor neurons. Neurons in the brain are called higher or central motor neurons because they are located in the central nervous system. Neurons in the brain stem or spinal cord are called lower or peripheral motor neurons.

Figure 1: Upper motor neurons in the brain and lower motor neurons in the brainstem and spinal cord .
the lower neurons of the brain stem and spinal cord
The upper motor neurons send electrical signals via cables to the lower motor neurons. These cables are long branches of the neuron, called axons. Together, these axons make up the corticospinal pathway in medical terms, as it runs from the cortex to the spinal cord, known as “medulla spinalis” in Latin. The electrical signals reach and activate the lower motor neurons. These in turn send their branches or axons to the muscles. These axons form the nerves that run from the spinal cord to the muscles. These axons are sometimes very long. While the neuron itself is only a tenth of a millimetre in diameter, an axon can easily be a metre long (i.e. 10,000 times longer). After all, it has to go from the spinal cord to the muscles of the foot, for example (Figure 2).
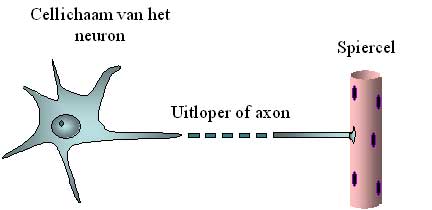
Figure 2: Schematic representation of the motor neuron cell body and its axon.
The axon is easily 10,000 times longer than the diameter of the cell body, which is why it is shown interrupted.
During the physical examination, the neurologist determines whether the motor neurons are functioning normally. EMG (short for electromyography) can identify abnormalities in peripheral neurons that are not even detectable on physical examination.
It is these two neurons that malfunction and later languish (‘degenerate’) in motor neurone disorders or motor neurone degeneration. Of these types of disease, ALS (amyotrophic lateral sclerosis) is the best known, but there are several types. Upper motor neuron malfunction mainly results in spasticity and abnormally sharp reflexes; lower motor neuron failure results in muscle weakness and atrophy. The latter term refers to the thinning of a muscle, clearly visible in the hand, for example.
In ALS, both types of neuron (central and peripheral) become diseased and die off. The result is a combination of symptoms: muscle wasting due to degeneration of the peripheral neurons, and at the same time abnormal reflexes and spasticity indicating degeneration of the central neurons. This combination, together with the continuous worsening of the disease, enables the doctor to diagnose ALS. The EMG will confirm that the peripheral neurons have been affected.
In the media, ALS and other motor neurone diseases are often described as “muscle diseases”. This is understandable, as muscle dysfunction is naturally the most visible. However, it is not the muscles that ‘go wrong’, but the nerve cells that have to supply them with the necessary signals.
In some patients, the disease is initially confined to the lower neurons. These people develop muscle atrophy and loss of strength, but no spasticity. In some of these patients, the signs of upper motor neurone disease then develop quite rapidly: they actually have ALS. However, another proportion of patients never develop signs of central motor neurone disease. Neurologists do not have an appropriate name for this disease. It is called lower motor neurone disease, or progressive spinomuscular atrophy (PSMA) or progressive muscular atrophy (PMA). This disease tends to progress more slowly than ALS, but this is not really well known. It is entirely possible that this form is in fact just a variant of ALS: after all, some people with a mutation in the SOD1 gene never develop spasticity and still have ALS.
A small proportion of patients develop only lower motor neuron abnormalities in the arms and upper motor neuron abnormalities in the lower limbs. This is known as “flail arm syndrome”. We know that this disease progresses quite slowly. It is only very late in its course that the muscles of speech and swallowing are affected.
The reverse is also true. In some patients, only the central motor neurons are affected. This form of motor neurone disease is known as primary lateral sclerosis (PLS). However, after a certain period of time, it becomes clear in some of these patients that the lower motor neurons are also affected: these people then have ALS. To detect the development of ALS, ALS patients undergo regular EMG tests.
The other part of the patient population never develops ALS. They really do have ALS. We don’t know much about this disease. We know that it is still 10 times less common than ALS. It is estimated that there are only around fifty ALS patients in Flanders. However, it is important to diagnose it correctly and to distinguish it from ALS. The outlook for PLS patients is different: the disease progresses much more slowly than ALS. A disease duration of ten or even fifteen years is not unusual for PDS, but it is for ALS.