
Présentation
La SLA est une maladie neurodégénérative progressive et finalement mortelle qui affecte les motoneurones responsables de l’initiation et du contrôle des mouvements musculaires volontaires et du diaphragme. La progression de la maladie entraîne une aggravation de la faiblesse et de l’atrophie musculaires, causant des difficultés à bouger les membres, à mâcher, à avaler, à parler et à respirer. Approximativement 30 000 personnes aux États-Unis vivent avec la SLA. Environ 90% des cas de SLA sont sporadiques, ce qui signifie qu’ils apparaissent sans antécédents familiaux clairs.
Malgré des antécédents familiaux négatifs, les personnes atteintes de SLA sporadique peuvent être porteuses de gènes associés à la maladie. Environ 10 % des personnes atteintes de SLA présentent une SLA familiale, généralement à transmission autosomique dominante. Des dizaines de gènes liés à la SLA ont été identifiés, notamment C9orf72, FUS, TARDBP et SOD1.
SOD1 a été le premier gène lié à la SLA et, les variantes associées à la maladie dans ce gène représentent 13 à 20 % des cas familiaux de SLA.
En avril 2023, le tofersen, commercialisé aux États-Unis sous le nom de Qalsody® (Biogen), a été approuvé par la FDA pour le traitement de la SLA liée à la SOD1. Le gène SOD1 code une protéine appelée superoxyde dismutase 1. Les mutations de la SOD1 liées à la SLA entraîneraient une forme « toxique » et mal repliée de la protéine, qui s’accumule dans les motoneurones, perturbe le métabolisme cellulaire et d’autres processus, et se propage entre les cellules. Le tofersen est un oligonucléotide antisens (ASO), un type de thérapie de précision conçu pour modifier l’expression d’un gène spécifique. Dans ce cas, le tofersen se lie à l’ARN messager (ARNm ou mRNA) de la SOD1 mutée et empêche la traduction en protéine, réduisant ainsi les niveaux de la protéine SOD1 mutée toxique.
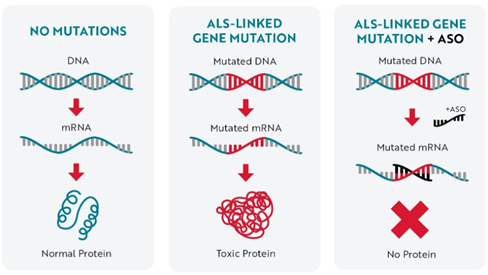
Diagramme illustrant les effets des mutations et du traitement par ASO sur l’expression génétique : une double hélice d’ADN normale, sans mutation, produit un ARN messager (ARNm) normal et une protéine saine ; une double hélice d’ADN présentant une mutation génétique liée à la SLA produit un ARNm muté et une protéine toxique ; un ASO se lie à l’ARNm muté, avec comme résultat la non production de protéines. Image credit: ALS Association.
Plus de 30 ans de recherche financée par la NIH ont contribué à des découvertes cruciales pour le développement du tofersen. En 1993, une étude financée par la NIH a établi que les variants de SOD1 étaient la première cause génétique connue d’un sous-ensemble de cas de SLA. Cette découverte a ouvert la voie à l’étude des mécanismes de la maladie et au testing de thérapies potentielles dans des modèles de souris génétiquement modifiés de SLA liée à SOD1 .Le développement du nusinersen (Spinraza), un traitement ASO pour l’amyotrophie musculaire spinale (AMS ou SMS), a démontré le potentiel de cette approche pour les maladies neurodégénératives (SMA). Des organisations universitaires américaines et internationales, des agences gouvernementales telles que la NIH et des entités privées ont investi dans la recherche d’une thérapie similaire pour la SLA liée à SOD1, ce qui a finalement conduit au développement du tofersen par Biogen et Ionis Pharmaceuticals.
Parallèlement, des recherches menées aux États-Unis et à l’étranger ont identifié une protéine structurelle distincte dans les neurones, appelée neurofilament à chaîne légère (NfL), comme biomarqueur fiable pour évaluer les lésions neuronales, la progression de la maladie et le pronostic de la SLA. L’approbation du tofersen par la FDA reposait sur la capacité du traitement à réduire les taux plasmatiques de NfL de 40-50 % sur une période de six mois. S’appuyant sur cette avancée, le premier essai de prévention de la maladie pour la SLA liée à SOD1 est actuellement en cours pour évaluer l’efficacité et la sécurité à long terme du tofersen. La NIH continue de soutenir la recherche visant à développer des ASO ciblant les gènes associés à la SLA et à explorer d’autres approches novatrices, telles que l’édition du génome, pour traiter la SLA.
Chronologie
1993
Des chercheurs établissent un lien entre des variantes du gène SOD1 et des cas familiaux de SLA.
1994
Des souris génétiquement modifiées avec un gène SLA humain muté suggèrent que la protéine mal repliée qui en résulte cause des dommages en s’accumulant dans les neurones.
1996
Des études montrent que les patients atteints de SLA et d’autres maladies neurodégénératives présentent des taux accrus de protéines de neurofilament à chaîne légère (NfL) dans le liquide céphalo-rachidien (LCR) – le liquide qui entoure et protège le cerveau et la moelle épinière. Ces résultats et d’autres suggèrent que le NfL pourrait être un marqueur significatif de la neurodégénérescence.
2003
Des chercheurs montrent que les cellules non neuronales exprimant la protéine SOD1 mutante contribuent significativement à la pathologie de la SLA. Cette découverte suggère que le ciblage de la protéine SOD1 mutante, de manière généralisée, à travers plusieurs types de cellules pourrait s’avérer une stratégie prometteuse pour le développement de thérapies.
2005
Dans un modèle de souris ALS, le silençage des gènes SOD1 mutés protège contre la neurodégénérescence et prolonge la survie.
2006
Le premier ASO conçu comme stratégie thérapeutique pour la SLA liée à SOD1 ralentit la progression de la maladie dans un modèle de rat.
2011
Une combinaison de biomarqueurs du CSF, y compris des protéines de neurofilament, semble prometteuses pour distinguer la SLA d’autres maladies neurodégénératives ou de cas témoins sains.
2015
Une étude clinique montre que les niveaux de neurofilament à chaîne légère (NfL) dans le plasma sanguin prédisent la progression de la maladie SLA, en s’appuyant sur des résultats antérieurs dans le CSF et en indiquant que les NfL dérivés du sang sont un biomarqueur plus accessible et moins invasif.
2016
Le premier essai clinique du tofersen commence.
2016
La FDA approuve le nusinersen (Spinraza) pour le traitement de l’AMS ou Atrophie Musculaire Spinale chez les enfants, démontrant le potentiel des thérapies ASO pour traiter les maladies neurodégénératives.
2018
Des chercheurs déterminent que la thérapie ASO dans les modèles de rongeurs atteints de SLA peut prolonger la survie et inverser la neurodégénérescence, même lorsque le traitement commence après le début de la maladie
2021
L’essai clinique de phase 3 du tofersen montre des réductions des niveaux de SOD1 et de NfL plasmatiques chez les participants traités, mais ne parvient pas à montrer une amélioration significative sur une mesure fonctionnelle de la progression de la maladie.
2023
La FDA accorde une autorisation accélérée au tofersen (Qalsody®) pour le traitement de la SLA SOD1, estimant que sa capacité à réduire les taux plasmatiques de NfL est susceptible de prédire un bénéfice clinique chez les patients. Des essais cliniques supplémentaires sont en cours pour évaluer la sécurité et l’efficacité à long terme du tofersen, notamment chez les personnes atteintes de variants SOD1 asymptomatiques.
Traduction: Viviane
Source: website ninds.nih.gov