

ALS, PLS, PMA: names with a meaning?
Prof. Dr. W. Robberecht
Neurology, UZ Gasthuisberg
Leuven
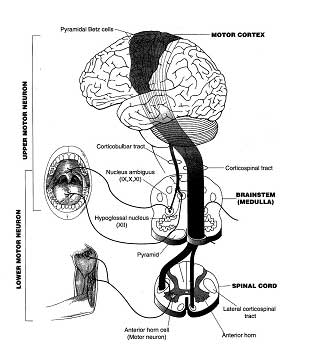
The planning of a movement happens in a series of complicated networks in the brain, of which the function goes wrong in the case of Parkinson’s disease or Huntington’s disease. To be able to execute a movement and generate muscle power, you have to activate two types of neurons. A first type of neurons is situated in the brain (in the motor gray substance, called the cortex); a second type in the spinal marrow to use the muscles in the arms, legs and trunk, or in the brainstem (the lower part of the brain) to use the swallow and speech muscles. Since these neurons guarantee the motor skills, they are called motor neurons. The ones in the brain are called the upper or central motor neurons, because they’re situated in the central nervous system. The ones situated in the brainstem or spinal marrow are called the lower or peripheral motor neurons.
![]()
Picture 1: The upper motor neurons in the brain
and the lower ones in the brainstem and the spinal marrow.
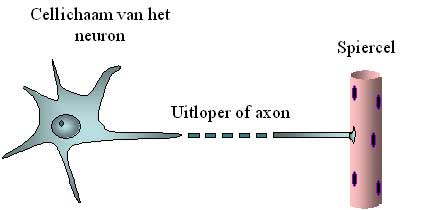
The upper motor neurons send electrical signals to the lower motor neurons through cables. These cables are long offshoots of the neuron, called axons. In medical terms, all these axons together are called the corticospinal tract, because they go from the cortex to the spinal marrow, called “medulla spinalis” in Latin. The electrical signals reach and activate the lower motor neurons. On their turn, the last ones send their offshoots or axons to the muscles. These axons form the neurons which go from the spinal marrow to the muscles. The axons can be very long: where the neuron is only a tenth of a millimetre in diameter, the axon can easily measure about a meter in length (10.000 times as long); since it has to go from the spinal marrow to, for example, the feet muscles (picture 2).
![]()
Picture 2 (cell organ of the neuron / offshoot or axon / muscle cell):
Schematic representation of the cell organ of the motor neuron and his axon.
The axon can easily be 10.000 times longer than the diameter of the cell organ and is therefore represented as a broken line.
While doing a physical examination, the neurologist can determine if the motor neurons are functioning normally. Using the EMG (abbreviation of electromyography), abnormalities of the peripheral neurons can be determined before they are traceable at a physical examination.
It’s these two neurons that malfunction and pine away (“degenerate”) in the case of motor neuron affections or motor neuron degenerations. Of this kind of diseases, amyotrophic lateral sclerosis is the most well-known, but there are several types. The malfunctioning of the upper motor neurons results in spasticity and abnormally vivid reflexes; the failing of the lower motor neurons results in muscle weakness and muscle atrophy. This last term means the thinning of a muscle, which is very visible at hand height.
In the case of amyotrophic lateral sclerosis (ALS), it’s the two types of neurons (the central and the peripheral) which fall ill and degenerate. This results in a combination of signs: the dissolving of the muscles by degeneration of the peripheral neurons, and abnormal reflexes and spasticity at the same time, which indicate the degeneration of the central neurons. This combination and the permanent deterioration of the disease, permit the doctor to determine ALS. The EMG will confirm the affection of the peripheral neurons.
The media describes ALS and other motor neuron affections often as “muscle diseases”. This is understandable since the malfunctioning of the muscles is the most visible symptom. Yet, it’s not the muscles that “make a mistake”, but the neurons which have to provide them with the required signals.
ALS occurs with approximately one or two people out of 100.000 a year. There are about 800 ALS-patients in Flanders. ALS occurs a little more often with men than with women. Most ALS-patients (90%) don’t have any family members with the same disease: this is what we call a sporadic or non-familial case. Approximately 10% of the patients do have family members with ALS and suffer from the familial form. From this form, we know the cause in about 20% of the cases (abnormalities in the SOD1 gene).
With some patients, the disease is in the beginning restricted to the lower neurons. These people develop muscle atrophy and loss of power, but no spasticity. Some of these patients quickly develop signs of a disease of the upper motor neuron: they do suffer from ALS. Another part of the patients never develop any signs of central motor neuron suffering. Neurologists don’t have a good name for this disease. It’s called lower motor neuron disease or progressive spinal muscular atrophy (PSMA) or progressive muscular atrophy (PMA). This disease tends to progress slower than ALS does, but this is not known for sure. It’s possible that this form is actually a variant of ALS: some people with an SOD1 mutation never develop spasticity but do have ALS by definition.
A small minority of the patients only develop abnormalities of the lower motor neurons in the arms and abnormalities of the upper motor neurons in the lower limbs. In English, it’s called the “flail arm syndrome”. It is known the disease develops rather slowly. It’s only very late in the evolution of this form that the muscles of the speech and swallow function are affected.
The opposite also exists. With some patients, only the central motor neurons are affected. This form of motor neuron disease is called primary lateral sclerosis (PLS). However, after a while, some of these patients also manifest an affection of the lower motor neurons: these people suffer from ALS. In order to detect the evolution to ALS, these PSL-patients often undergo an EMG.
The other part of these patients never develop this evolution to ALS. They really suffer from PLS. There’s not much known about this disease. We know it occurs about 10 times less than ALS. It is estimated that there are only about 50 PLS-patients in Flanders. However, it’s important to diagnose correctly, and to make the difference with ALS. The prospects of PLS-patients are different: the disease develops much slower than ALS. A disease duration of ten or even fifteen years isn’t unusual for PLS, but it is for ALS.
The health insurance repays riluzole only to people who meet all the requirements of ALS. A lot of these patients don’t meet these requirements in the beginning of the disease, and wouldn’t be repaid the medicine. This means a loss of time: after a few months or even a year, they’re only showing all the symptoms the Belgian Institute of Illness and Invalidity Insurance (RIZIV) requires. However, many ALS-specialists think riluzole has mainly an effect in the beginning of the affection.
The fact that RIZIV doesn’t pay back riluzole to people who only have signs of abnormalities of the lower motor neurons is regrettable, because these patients almost always have a form of ALS, as we mentioned before. In the case of PLS, the health insurance doesn’t pay back riluzole either. This is understandable. The development of the disease progresses slow enough to presume that the medicine won’t be useful. We don’t know it for sure. The Netherlands have developed a study which tries to investigate this matter. The result seems predictable.