

SLA, SLP, AMP : que se cache-t-il derrière ces noms?
Prof. Dr W. Robberecht
Neurologie, UZ Gasthuisberg
Louvain
La planification d’un mouvement s’effectue dans un ensemble de réseaux très complexes du cerveau, dont le fonctionnement perturbé est à l’origine de maladies comme celle dit de Parkinson ou de Huntington. Pour aboutir à l’exécution d’un mouvement et donc pour générer de la force musculaire, deux types de neurones doivent être activés. Le premier type de neurone est situé dans le cerveau (dans la substance grise motrice, appelée écorce ou cortex), alors que le second type se trouve dans la moelle épinière pour activer les muscles des bras, des jambes et du tronc, ou dans le cervelet (partie inférieure du cerveau) pour commander les muscles de la déglutition et de la parole. Comme ces neurones sont responsables de la motricité, on les appelle motoneurones. Ceux qui se trouvent dans le cerveau sont désignés par le terme motoneurones supérieurs ou centraux, parce qu’ils font partie du système nerveux central. Ceux du cervelet ou de la moelle épinière portent le nom de motoneurones inférieurs ou périphériques.
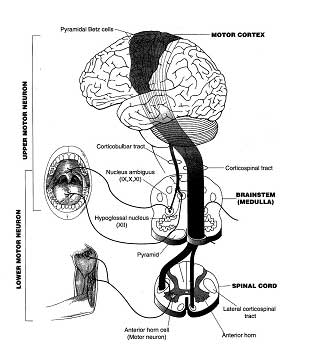
Illustration 1: Les motoneurones supérieurs dans le cerveau,
les motoneurones inférieurs dans le cervelet et la moelle épinière
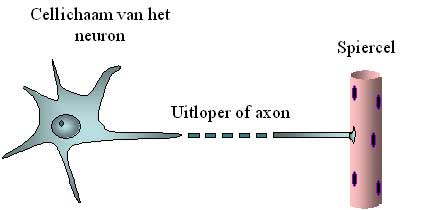
Les motoneurones supérieurs envoient des signaux électriques aux motoneurones inférieurs par l’intermédiaire de câbles. Ces câbles ou axones sont de longs prolongements des neurones. L’ensemble de ces axones est désigné en termes médicaux comme faisceau corticospinal parce qu’il relie le cortex à la moelle épinière (en latin, « medulla spinalis »). Les signaux électriques atteignent et activent les motoneurones inférieurs, dont les prolongements ou axones sont à leur tour reliés aux muscles. Ces axones forment les nerfs qui relient la moelle épinière aux muscles. Ils sont parfois très longs : alors que le diamètre d’un neurone ne mesure qu’un dixième de millimètre, un axone peut facilement atteindre une longueur d’un mètre (donc 10.000 fois plus long) ; il doit en effet relier la moelle épinière à, par exemple, les muscles du pied. (Illustration 2)
![]()
Illustration 2: Représentation schématique d’un noyau cellulaire de neurone moteur et de son axone.
L’axone, qui peut facilement être 10.000 fois plus long que le diamètre de la cellule, est figuré par un pointillé.
Lors d’un examen physique, le neurologue peut vérifier si les motoneurones fonctionnent normalement. L’EMG (acronyme de électromyographie) permet de déceler des anomalies des neurones périphériques qui ne sont pas détectables lors d’un examen physique normal.
Ce sont ces deux neurones qui fonctionnent mal et dépérissent (« dégénèrent ») par la suite à cause d’affections des motoneurones ou de dégénérescences des motoneurones. Parmi les maladies de ce type, la plus connue est la sclérose latérale amyotrophique, mais il en existe différentes autres. Un mauvais fonctionnement des motoneurones supérieurs engendre essentiellement la spasticité et des réflexes anormalement vifs, alors qu’une déficience des motoneurones inférieurs provoque une faiblesse musculaire et une atrophie musculaire. Cette dernière signifie que le muscle s’amenuise, comme on peut par exemple très bien le remarquer au niveau de la main.
La sclérose latérale amyotrophique (SLA) atteint et fait dépérir les deux types de neurones (le central aussi bien que le périphérique). Elle engendre une combinaison de symptômes : amenuisement des muscles dû à la dégénérescence des neurones périphériques, et apparition simultanée de reflexes anormaux et de spasticité qui indiquent la dégénérescence des neurones centraux. Cette combinaison et le fait que la maladie empire constamment permettent au médecin de diagnostiquer la SLA. Une EMG confirmera l’atteinte des neurones périphériques. En parlant de SLA et d’autres affections des motoneurones, les médias utilisent souvent le terme de « maladies musculaires ». C’est compréhensible parce que le mauvais fonctionnement des muscles est évidemment le symptôme le plus apparent. Ce ne sont pourtant pas les muscles qui sont en cause, mais les cellules nerveuses qui doivent leur envoyer les signaux appropriés. La SLA atteint environ 1 à 2 personnes sur 100.000 par an. La Flandre compte à peu près 800 patients atteints de SLA. La SLA est légèrement plus fréquente chez les hommes que chez les femmes. La majorité des patients frappés par la SLA (90%) n’ont dans leur famille aucun membre souffrant de la même maladie : nous les désignons comme cas sporadiques ou non familiaux. Environ 10% des patients ont par contre bel et bien des membres de la famille atteints de SLA et souffrent donc de la forme familiale de celle-ci. La cause de cette forme familiale de la SLA est connue dans à peu près 20% des cas (anomalies du gène SOD1).
Chez certains patients, la maladie n’atteint au début que les neurones inférieurs. Ces personnes développent une atrophie musculaire et souffrent de perte de puissance, mais pas de spasticité. Parmi eux, il y en a qui présentent néanmoins assez rapidement des symptômes indiquant une affection des motoneurones supérieurs. Ils sont alors vraiment atteints de SLA. D’autres ne développent par contre jamais de signes de maladie des motoneurones centraux. Jusqu’à présent, les neurologues n’ont pas encore trouvé de nom approprié pour cette maladie. Ils parlent d’une maladie des motoneurones inférieurs ou d’atrophie spinomusculaire progressive (ASMP) ou d’atrophie musculaire progressive (AMP). Cette maladie a tendance à évoluer plus lentement que la SLA, mais on n’a pas de certitude. Il se pourrait très bien que cette forme ne soit en réalité qu’une variante de la SLA, car certaines personnes atteintes de la mutation du SOD1 ne souffrent eux non plus jamais de spasticité alors qu’elles sont par définition quand même atteintes de SLA. Un petit nombre de patients développent uniquement des anomalies des motoneurones inférieurs dans les bras et uniquement des anomalies des motoneurones supérieurs dans les membres inférieurs. En anglais, on parle de “flail arm syndrome”. On sait que l’évolution de cette maladie est plutôt lente et qu’elle n’affecte les muscles de la fonction de la parole et de la déglutition que lorsqu’elle a atteint un stade très avancé.
L’inverse existe également. Chez certains patients, seuls les motoneurones centraux sont affectés. Cette forme de maladie des motoneurones est appelée sclérose latérale primaire (SLP). Après un temps, on peut toutefois constater que les motoneurones inférieurs de certains de ces patients sont eux aussi manifestement touchés. Ces gens ont alors quand même la SLA. Pour déceler une évolution vers la SLA, on fait régulièrement subir une EMG aux patients qui souffrent de SLP. Les autres patients ne développent jamais d’évolution vers la SLA. Ils sont donc vraiment atteints de SLP. Cette maladie est mal connue. On sait qu’elle est à peu près 10 fois moins fréquente que la SLA. En Flandre, on évalue à une cinquantaine le nombre de patients atteints de SLP. Il est néanmoins important d’établir un diagnostic exact et de distinguer cette maladie de la SLA. Les perspectives d’avenir des patients qui souffrent de la SLP sont en effet différentes, car la SLP évolue beaucoup plus lentement que la SLA. Un patient atteint de SLP peut très bien survivre pendant 10 à 15 ans, ce qui est tout à fait inhabituel pour une personne souffrant de SLA.
L’assurance maladie ne rembourse la riluzole qu’à des malades qui répondent à tous les critères de la SLA. Or, comme beaucoup de patients ne répondent en début de maladie pas à tous ces critères, l’achat de leur médicament ne serait pas remboursé. Ceci constitue en fait une perte de temps, puisqu’ils ne répondront à tous les critères exigés par l’INAMI que quelques mois, voire un an, plus tard. De nombreux spécialistes de la SLA estiment pourtant que la riluzole est surtout efficace en début de maladie. Il est dès lors regrettable que l’INAMI ne rembourse pas la riluzole à des personnes qui présentent uniquement des symptômes d’anomalies au niveau des motoneurones inférieurs, car ces patients sont, comme nous l’avons signalé, pratiquement toujours atteints d’une forme de SLA. En cas de SLP, l’assurance maladie refuse également de rembourser la riluzole. Ce qui est compréhensible. L’évolution de cette maladie est en effet tellement lente qu’on est en droit de supposer que le médicament ne sera d’aucune utilité. Ce qui est loin d’être certain. Aux Pays-Bas, on a entamé une étude qui essaiera d’examiner ce problème. Les résultats de cette étude semblent tout à fait prévisibles.