

Laten we familiale ALS vanaf vandaag verhelpen
07-07-2016
 Onze gezondheidsinstanties zijn in staat familiale ALS uit de wereld te helpen. We beschikken daarvoor over de nodige technologie.
Onze gezondheidsinstanties zijn in staat familiale ALS uit de wereld te helpen. We beschikken daarvoor over de nodige technologie.
Is de wereld echter klaar om deze technologie te aanvaarden en ze in te schakelen om veel families van deze verwoestende ziekte te verlossen?
In 2008 kreeg Debra Quinn voor het eerst last van zwakte in haar rechterbeen. Een jaar later werd ze formeel gediagnosticeerd met ALS. Dat kwam niet als een verrassing voor Debra; de ziekte teistert haar familie al heel lang. Haar grootmoeder Dora, haar tante Bonnie, haar vader Bill en haar zuster Rhonda kregen allemaal ALS en stierven eraan, een tragische familiegeschiedenis, als we de genealogie in ogenschouw nemen. Een genealogie is als een stamboom die je kunt opmaken via Ancestry.com. De genealogie van de familie Quinn verschilt niet van die van andere families met familiale ALS — een geschiedenis van terminale ziekte die vele generaties teruggaat.
In tegenstelling tot de meeste ziektes bestaan er twee vormen van ALS.
Sporadische ALS doet zich voor wanneer de onderliggende genetische tekortkoming of omgevingsfactor grotendeels onduidelijk blijft, en wanneer de ziekte niet wordt 'doorgegeven' van generatie op generatie via mendeliaanse genetische transmissie. Tot 90% van alle ALS-gevallen zijn sporadisch.
Jammer genoeg is de familie Quinn een geval van familiale ALS (fALS), een ziektevorm die van generatie op generatie wordt overgeërfd. Genealogieën die honderden jaren teruggaan brengen de overerving van de ziekte van ouders naar kinderen in kaart. Bij veel families is de overerving erg dominant. Dat betekent dat één of meerdere kinderen vaak de genetische voorbestemming erven en uiteindelijk gediagnosticeerd worden met ALS.
Debra Quinn heeft te lijden van de vreselijke initiële ongemakken waarmee alle ALS-patiënten kampen. Ze zit vaak in een rolwagen en kan alleen erg korte afstanden te voet aan. Haar familie zal binnenkort een traplift plaatsen om Debra de kwelling te besparen een trap te moeten opgaan. Ook met de meest elementaire dagtaken heeft ze het moeilijk.
Wat de tragedie nog erger maakt, is dat Debra twee relatief jonge kinderen heeft: haar zoon Dustin is 28 en haar dochter Kristin 29. Beiden hebben ze weet van hun ALS-familiegeschiedenis. Ze zijn ervan op de hoogte dat hun familie een mutatie in een gen met de naam superoxide dismutase 1 (SOD1) doorgeeft.
In 2013 onderging Dustin tests. De resultaten waren positief voor de SOD1-mutatie in zijn familie. Kristin onderging de test in 2014 en de resultaten bleken negatief voor mutaties in het SOD1-gen. De toekomst ziet er dus niet rooskleurig uit voor een mooie familie waarvan de moeder de ziekte al heeft en de zoon ze dreigt te krijgen, al zal de dochter dit lot wellicht bespaard blijven.
Het heeft tientallen jaren gekost om de hogedoorvoersequentiëringstechnologieën te ontwikkelen die de sequentiëring van de eerste voorlopige versie van het menselijk genoom mogelijk maakten. Gedurende die periode gebruikten wetenschappers 'brute kracht'-mappingtechnieken om de ziekteveroorzakende genetische modificaties te identificeren. Het mag geen verrassing heten dat het bijna 200 jaar heeft geduurd voor we van de eerste beschrijving van een motorneuronenziekte door Charcot uitkwamen bij de mapping en sequentiëring van de eerste genetische mutatie geassocieerd met ALS. Een mutatie in het gen dat het enzym superoxide dismutase 1 (SOD1) encodeert en dat werd vermeld in een publicatie in Science in 1993 (Rosen), staat in voor ongeveer 2% van alle ALS-gevallen.
Naarmate DNA-sequentiëringstechnologieën zich hebben zich ontwikkeld met het tempo van de Wet van Moore, zijn de kosten gereduceerd om grotere cohorten patiënten te sequentiëren, en zijn tientallen andere met ALS geassocieerde genen geïdentificeerd, zoals TDP43, C9ORF72, FUS1 en nog vele andere.
En nu steeds meer families worden gesequentieerd, is duidelijk geworden dat er voor veel van de genen meerdere en verschillende mutaties bestaan die kunnen leiden tot ALS.
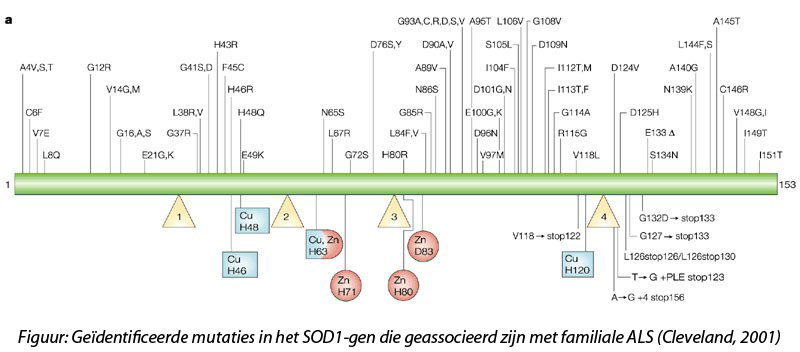 In de nabije toekomst zal de wetenschappelijke gemeenschap alle genen die geassocieerd zijn met familiale vormen van ALS hebben geïdentificeerd.
In de nabije toekomst zal de wetenschappelijke gemeenschap alle genen die geassocieerd zijn met familiale vormen van ALS hebben geïdentificeerd.
Ondanks al die vergaarde kennis van de genetica van de ziekte, blijft de gemeenschap van ALS-patiënten echter nog altijd verstoken van efficiënte behandelingen, zelfs de kleine groep die het slachtoffer is van mutaties in SOD1-families, zoals de familie Quinn. En dan te weten dat de groep mutaties die aan de basis ligt van hun vorm van de ziekte al meer dan 20 jaar geleden werd geïdentificeerd!
En laat ons wel wezen, ook voor patiënten met andere gekende vormen van genetische mutaties bestaan geen efficiënte behandelingen, en voor sporadische ALS evenmin.
Maken wetenschappers, clinici, ondernemingen en andere belanghebbenden dan geen werk van doelgerichte therapieën om deze specifieke 'genetische subtypes' van ALS te behandelen?
Natuurlijk wel.
Er bestaan diverse ontwikkelingsstrategieën om veel van deze genetische vormen van ALS aan te pakken. Sommige invalshoeken doen een beroep op decennia van vooruitgang op het vlak van gentherapie of antisense-RNA-technologieën.
Andere richten zich meer op traditionele vormen van geneesmiddelenontwikkeling, door zich toe leggen op eiwitbiologics, met als doelwit muterende vormen van de eiwitten die worden geëncodeerd door deze genetische mutaties.
Bovendien ben ik ervan overtuigd dat er voorstellen zullen worden gedaan om genetische editing tools te gebruiken (Zinc Finger Nucleases, Talens, en CRISPRs) om de genetische abnormaliteiten die aanleiding geven tot ALS te herstellen.
Aangezien echter de klinische ontwikkeling van deze invalshoeken vooralsnog uitblijft wat ALS betreft, kan het nog een tiental jaar duren voor een efficiënte behandeling voorhanden is voor ALS-patiënten. Bovendien moet voor elk soort aangetast gen een verschillende therapie worden ontwikkeld en mogelijk is er zelfs behoefte aan verschillende therapieën voor de diverse subtypes van mutaties in één en dezelfde genfamilie, wat tientallen jaren in beslag kan nemen.
Dat is naar mijn mening te lang om het grootste kapitaal op deze planeet verloren te laten gaan: mensenlevens en hun invloed op hun geliefden, families zoals die van Debra, Kristin, en Dustin Quinn.
We kunnen echter vandaag nog beginnen met het uitroeien van familiale ALS!
Hoe dan?
Door genetische screening gekoppeld aan in-vitrofertilisatie (IVF)!
Dit idee is niet nieuw. De combinatie van deze twee technologieën werd al toegepast bij andere indicaties van ernstige genetische aandoeningen en lag aan de basis van een totaal nieuw medisch vakgebied met de naam Pre-implantatie Genetische Diagnose (PGD).
Bij PGD gaat het om de in-vitrofertilisatie van een eicel met het sperma van de partner. Drie dagen na de bevruchting, tijdens het blastomerenstadium, wordt één enkele cel verwijderd en wordt een polymerasekettingreactie gebruikt om het potentieel gemuteerde gen, zoals SOD1 of TDP43, te vergroten. Alleen bevruchte eicellen die de potentiële mutatie niet bevatten worden in de uterus ingeplant. Als gevolg hiervan dragen nakomelingen niet langer de genetische mutatie die geassocieerd wordt met familiale ALS en lopen ze niet langer het risico het gemuteerde gen door te geven aan hun eigen nageslacht. Op die manier wordt de cyclus van familiale ALS voorgoed doorbroken!
Om iedereen op zijn gemak te stellen, wil ik hieraan toevoegen dat hier geen sprake is van genetische manipulatie in de gangbare betekenis. Geen stamcellen dus, geen modificatie van het genoom, geen andere oogkleur.
Als je denkt dat je de drager bent van een mutatie, zonder dat je hiervan op de hoogte wilt zijn, is het trouwens ook mogelijk voor een PGD-procedure te kiezen om uit te maken of je kinderen de mutatie hebben, zonder dat je over je eigen status wordt ingelicht.
En toch, terwijl ik dit bericht schrijf, zul je op PubMed voor de zoekterm 'amyotrophic' gecombineerd met 'IVF' of 'PGD' geen enkel artikel vinden!
Dat was een compleet raadsel voor mij. Waarom bestaat er geen intens actieve wetenschappelijke en klinische strategie om families met familiale ALS aan te moedigen systematisch PGD te gebruiken? Een goed uitgevoerd plan om PGD te implementeren kan ALS genezen bij 10% van de ziektepopulatie. Dat biedt een grotere garantie op succes dan veel geneesmiddelen die goedgekeurd zijn door het FDA (Voedsel- en Geneesmiddelenagentschap van de VS).
IVF is een wereldwijd gangbare medische procedure die wordt toegepast om de levenskwaliteit te verbeteren van vrouwen die een kind willen in omstandigheden waarbij conceptie problematisch blijkt. Het is een procedure die een mooi voorbeeld vormt van de stapsgewijze vooruitgang die vaak vereist is voor niet alleen medische procedures, maar ook voor de ontwikkeling van bijna alle medische behandelingen.
In 1977 onderging Leslie Brown in Engeland de eerste IVF-procedure. Ze kreeg een gezonde baby: Louise. Momenteel worden in de VS jaarlijks naar schatting meer dan 60.000 succesvolle IVF-procedures toegepast en wereldwijd werden al meer dan 5 miljoen succesvolle IVF-procedures uitgevoerd!
De gemiddelde kostprijs van IVF bedraagt tussen de 15.000 en 20.000 euro, met een succesratio van meer dan 40% vóór de leeftijd van 35.
Ik moet hier wel bij vermelden dat IVF fysiek en emotioneel geen lachertje is, vooral voor de vrouw. We hebben het hier echter over het vermijden van ALS en dat zegt volgens mij genoeg.
Bij meer dan 300 andere ziekte-indicaties had PGD een ongelooflijke impact. Nemen we het voorbeeld van Amanda and Bradley Kalinsky. Amanda heeft de genetische mutatie voor de ziekte van Gerstmann-Straussler-Scheinker (GSS). Dankzij PGD heeft het gezin nu drie gezonde kinderen zonder de genetische mutaties die GSS veroorzaken.
De kinderen van Amanda zullen een gezond leven leiden en kunnen er zeker van zijn dat ze het muterende gen nooit zullen doorgeven aan hun eigen kinderen, ook al zal Amanda uiteindelijk een ziekte krijgen die enigszins op ALS lijkt en ongeveer vijf jaar na het intreden van de symptomen fataal kan zijn.
Waarom is PGD dan niet hét gespreksonderwerp tijdens jaarlijkse ALS-bijeenkomsten? Aan de kostprijs of de mate van succes kan het zeker niet liggen. Laten we eens kijken naar aanverwante indicaties om dit raadsel enigszins te ontcijferen.
Er werd een onderzoek uitgevoerd bij huisartsen, neurologen en psychiaters met de vraag of ze PGD zouden aanraden bij een patiënt met de ziekte van Huntington (ZH), een patiënt met Cystische Fibrose (CF), of een patiënt die een niet-dodelijke selectietest wil om het geslacht van zijn kind te bepalen. De resultaten zijn verrassend.
Bij een onderzoek waarbij 163 neurologen en 372 psychiaters betrokken waren, had slechts 2,9% PGD besproken met patiënten met een genetische afwijking. Daartegenover staat dat 24,9% van de neurologen en 31,9% van de psychiaters prenatale genetische tests van foetussen hadden besproken met patiënten die drager waren van genetische aandoeningen (Klitzman,2013; Klitzman, 2014).
Toen hen echter werd gevraagd of ze PGD zouden aanraden aan patiënten met de ziekte van Huntington of met Cystische Fibrose, of aan mensen die een selectieve geslachtstest wilden, antwoordden de meeste artsen 'ja' als het erom ging PGD te bespreken met hun patiënten die positief scoorden op ZH en CF, maar waren ze heel wat terughoudender om de procedure toe te passen voor geslachtskeuze. Deze gegevens suggereren dat gespecialiseerde artsen beter vertrouwd zijn met PGD en bereid zijn dit te bespreken met patiënten die er hun voordeel mee kunnen doen en het daarom in overweging zouden kunnen nemen.
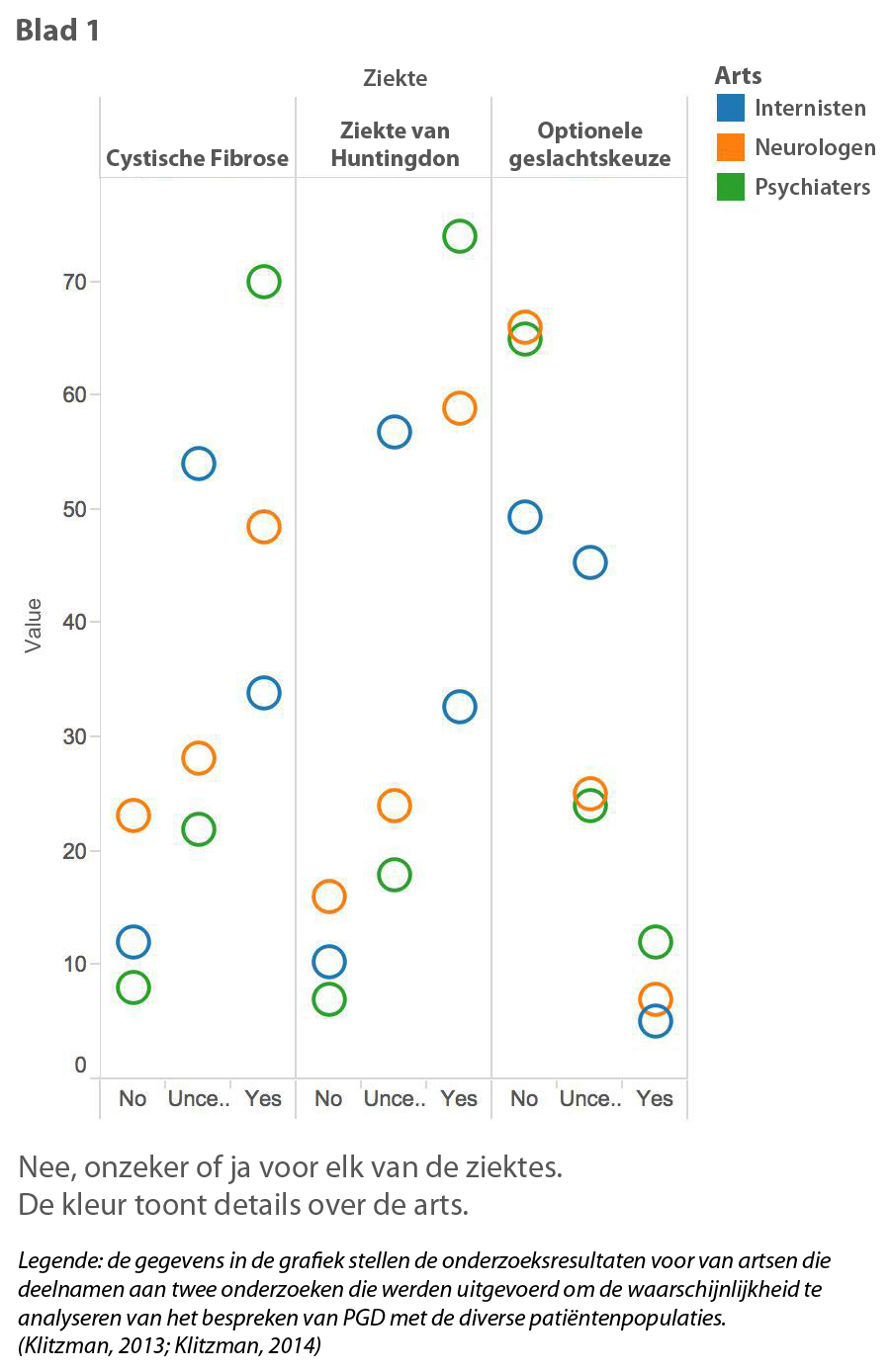 Als we naar deze gegevens kijken, is het niet moeilijk vast te stellen dat ALS-neurologen zich ongemakkelijk of onzeker voelen over de mogelijke toepassing van PGD op familiale ALS-patiënten in hun hospitaal. Belangrijker nog, ze zijn niet vertrouwd met het aangetoonde succes bij ziektes zoals ZH.
Als we naar deze gegevens kijken, is het niet moeilijk vast te stellen dat ALS-neurologen zich ongemakkelijk of onzeker voelen over de mogelijke toepassing van PGD op familiale ALS-patiënten in hun hospitaal. Belangrijker nog, ze zijn niet vertrouwd met het aangetoonde succes bij ziektes zoals ZH.
Het succes van het proces bij ZH is eigenlijk ongelooflijk, zelfs zonder wijdverbreide kennis van en discussie over de potentiële toepassing ervan op verwoestende neurologische ziektes.
Gegevens van een Europees consortium van meerdere centra dat zich bezighoudt met ZH geven het volgende beeld:
"In het totaal startten 257 paren met de voorbereiding en 174 paren (70% via een directe test, 30% via een exclusieve test) maakten op zijn minst één PGD-cyclus door. In het totaal werden 389 cycli voortgezet tot de eicelvergaring (EV). De bevallingsratio's per EV bedroegen 19,8%, en per embryotransfer 24,8%, met als resultaat 77 bevallingen en de geboorte van 90 kinderen." (Rij, 2012)
De volgende generatie van deze families is voorgoed van ZH verlost. We kunnen ons de impact hiervan op ALS-families zo al voorstellen. Als we nog maar een handvol families verlossen van familiale ALS, zou dat de grootste doorbraak zijn op het vlak van ALS sinds de ontdekking van de SOD1-genmutatie in 1993.
Volgens een recent verslag gepubliceerd door The Muscular Dystrophy Association (Musculaire Dystrofievereniging), bedraagt de jaarlijkse kostprijs voor het verzorgen van ALS-patiënten in de VS bijna 1,1 miljard dollar (of euro) (Larkindale, 2014). Als via PGD ook maar een fractie van de familiale ALS-gevallen kan worden voorkomen, zou dat de economie miljarden dollars besparen in de volgende paar generaties.
Is de wereld hier echter klaar voor?
Uiteraard denk ik hier aan de ethische bezwaren inzake PGD. Er bestaan tientallen studies en opvolgingsverslagen over de discussie omtrent PGD en de toepassing ervan op patiënten. Deze studies hebben de rechtvaardiging van PGD proberen te behandelen in verschillende gevallen, zoals hoogpenetrante kinderziektes, hoogpenetrante ziektes die zich bij volwassenen voordoen, ziektes waarbij de genetische vatbaarheid niet zo duidelijk is, en de selectie van eigenschappen die niet geassocieerd zijn met ziekte.
Uit deze studies blijkt duidelijk dat de toepassing van PGD voor de selectie van kenmerken die niet aan ziekte gekoppeld zijn geen ethisch draagvlak heeft bij de medische gemeenschap en ook niet bij andere gemeenschappen.
Voor ziektes waarbij een genetisch kenmerk mogelijk nooit een ziekte tot gevolg heeft (lage penetratie), staat het ten zeerste ter discussie in welke gevallen PGD mag worden besproken met de patiënt of hem mag worden aangeraden.
Voor hoogpenetrante kinder- en volwassenenziektes waarvoor geen efficiënte behandeling bestaat, wordt PGD als zeer aanvaardbaar beschouwd door de medische gemeenschap.
In één van deze verslagen, van het Ethics Committee of the American Society for Reproductive Medicine (Ethisch Comité van de Amerikaanse Vereniging voor Reproductieve Geneeskunde) staat te lezen:
"Pre-implantatie genetische diagnose (PGD) voor aandoeningen die zich bij volwassenen voordoen is ethisch gerechtvaardigd in ernstige omstandigheden en als er geen mogelijke interventies gekend zijn voor deze aandoeningen of als de beschikbare interventies ofwel niet efficiënt genoeg zijn ofwel aanzienlijk belastend."
Als dit niet strookt met de definitie van ALS, weet ik het ook niet meer.
De ethische en religieuze discussies over de gepaste implementatie van PGD liggen duidelijk buiten het bereik van deze discussie en er bestaan waarschijnlijk geen pasklare antwoorden. Alles hangt af van de individuele ouders of families. Zij moeten kiezen voor het minste kwaad: een verwoestende ziekte doorgeven aan je kinderen zonder dat een efficiënte behandeling mogelijk is of de morele verplichting een bevruchte eicel ongemoeid te laten die een gekende genetische mutatie bevat.
Op dit punt gekomen, zou de hamvraag in een quizprogramma als volgt kunnen luiden: "Als jij of je vrouw ALS hadden, zouden jullie dan van PGD gebruikmaken?"
Voor mij is dat een uitdagende vraag, aangezien ALS niet voorkomt in mijn familie. Anderzijds had ik wel vier ooms langs vaderszijde die na hun veertigste stierven aan een myocardiaal infarct door toedoen van een geblokkeerde slagader. Mijn vader is een kranige tachtiger, maar ikzelf kreeg in 2010 af te rekenen met een ernstige slagaderblokkering zonder voorafgaande tekenen. Ik schoot er bijna het leven bij in, ware het niet dat er op die bewuste ochtend om 7 uur een collega-TDI-wetenschapper van de vroege shift in het lab aanwezig was. Ik dankte mijn leven aan zijn reanimatie en aan het feit dat het Mass General Hospital op maar 5 minuten afstand lag. Ik heb twee jonge, mooie dochters en ik weet niet of ik de vatbaarheid van mijn familie voor cardiovasculaire ziektes aan mijn kinderen heb doorgegeven, maar uiteraard hoop ik van niet. Als ik de mogelijkheid had gehad om deze vatbaarheid te laten testen door middel van een PGD-test, zou ik geen seconde getwijfeld hebben.
Ik wil alle neurologen aanmoedigen om families met familiale ALS in te lichten over de mogelijkheden van PGD. Ik wil ook alle ALS-organisaties aanmoedigen deze strategie aan de nodige fondsen te helpen. Samen kunnen we bijdragen aan het verhelpen van familiale ALS terwijl we op zoek blijven gaan naar efficiënte behandelingen voor alle ALS-patiënten.
Laten we vandaag nog beginnen met de uitroeiing van familiale ALS! Echt waar, vandaag nog …
Als gemeenschap kunnen we de nodige inlichtingen verschaffen en steun en begeleiding bieden aan families met SOD1, TDP43, C9ORF72, en alle andere gekende familiale mutaties.
Geen enkele ziekte kun je zomaar van dag op dag uit de wereld helpen, dus laten we vandaag nog beginnen met de uitroeiing van familiale ALS.
Steve (Perrin CEO ALS TDI)
Vertaling: Bart De Becker
Bron: Translate ALS