

Forme mystérieuse de SLA identifiée chez des enfants présentant des mutations génétiques rares
06-06-2021
La SLA est une maladie neurologique caractérisée par une dégénération des motoneurones entraînant la perte des mouvements musculaires volontaires.
La condition, provoquant une faiblesse croissante des muscles à travers le corps, rend difficiles la marche, la parole et finalement même la respiration, entraînant la mort dans la plupart des patients à peine quelques années après l’apparition des premiers symptômes.
La majorité des cas de SLA apparaissent chez des personnes âgées de 55 à 75 ans, et la plupart des cas sont considérés comme sporadiques, la cause restant finalement inconnue.
Or, chez certaines personnes, la maladie se présente très différemment. Dans des cas rares, la génétique semble jouer un rôle causal – la maladie apparaissant parfois chez des personnes beaucoup plus jeunes.
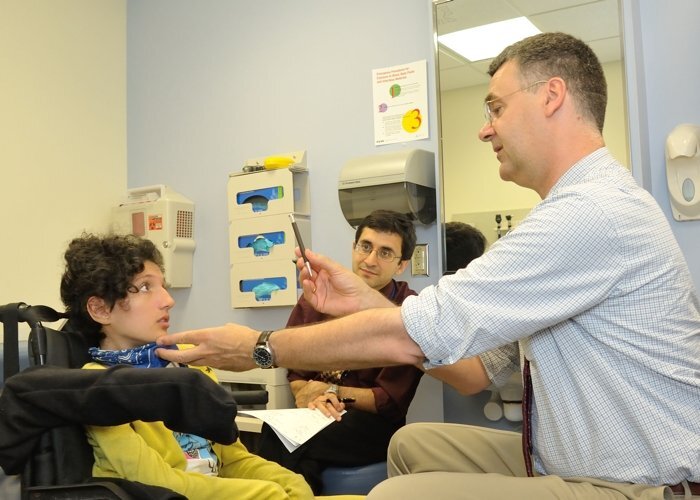
Image: Les auteurs de l’étude Carsten Bönnemann (à droite) et Payam Mohassel (à gauche) évaluent la patiente Claudia Digregorio.
Dans une nouvelle étude, les scientifiques ont pu démontrer la coïncidence de ces manifestations plus rares, ayant découvert une série inhabituelle de mutations liée à une forme distincte de SLA génétique identifiée chez des enfants, dont les maladies atrophiant les muscles ont occupé leurs médecins pendant des années.
Dans le plus médiatisé de ces cas poignants, une adolescente italienne Claudia Digregorio a fini par rencontrer le pape après que sa maladie dégénérative non identifiée avait attiré l’attention sur Youtube.
En même temps, des scientifiques à l’Institut national des troubles neurologiques et des accidents vasculaires cérébraux (NINDS) à Bethesda, Maryland ont commencé à étudier la maladie de Claudia, dans l’espoir de d’en découvrir la cause sous-jacente.
Dans une étude internationale dédiée à 10 jeunes patients répondant à ces critères – beaucoup d’entre eux ont commencé à développer des symptômes similaires à ceux de la SLA dès la petite enfance – des chercheurs ont pu identifier une base génétique pour ce sous-type rare d’une maladie déjà rare en soi.
« Ces jeunes patients souffraient d’une bonne partie des troubles des motoneurones supérieurs et inférieurs caractéristiques de la SLA », explique le neurologue Payam Mohassel des Instituts américains de la santé (NIH).
« L’aspect unique de ces cas est représenté par l’âge précoce d’apparition ainsi que par la lente progression des symptômes. Ces éléments nous ont amenés à chercher la cause sous-jacente de cette forme distincte de SLA. »
La réponse semble résider dans une série de mutations appelée SPTLC1, qui encode une protéine agissant comme catalyseur dans la production de molécules grasses appelées sphingolipides.
L’ADN des 10 jeunes patients présentait des mutations dans le gène SPTLC1, quatre des patients (d’une même famille) ayant hérité leurs variations d’un de leurs parents, tandis que les six autres, nullement unis par un lien de famille, présentaient des mutations de novo aléatoires (se manifestant pour la première fois chez un membre de la famille) dans le gène.
Dans les deux cas, les mutations se révèlent problématiques, menant à des productions excessives de sphingolipides, liées à des concentrations anormalement élevées d’une enzyme aidant à produire des lipides, appelée Sérine palmitoyltransférase (SPT).
« Nos résultats suggèrent que ces patients souffrant de SLA vivent essentiellement sans interruption de l’activité SPT », explique la biochimiste Teresa M. Dunn de la Uniformed Services University
« La SPT est contrôlée par une boucle de rétroaction… Les mutations que présentent ces patients court-circuitent essentiellement cette boucle de rétroaction. »
Des anomalies dans l’activité SPT dues aux mutations dans le gène SPTLC1 ont précédemment été attribuées à la neurodégénérescence causée par une autre maladie appelée Neuropathie héréditaire sensitive et autonomique (HSAN1), quoique le mécanisme pathologique s’avère très différent.
En cas de HSAN1, les mutations du gène SPTLC1 produisent des sphingolipides nocifs; dans la présente étude, les mutations du SPTLC1 trouvées produisaient des quantités anormales de sphingolipides non nocives, en empêchant une protéine appelée ORMDL de réguler l’activité SPT.
À son tour, l’excédent de sphingolipides s’accumule dans les motoneurones humains, entraînant la forme précoce de la maladie du motoneurone, que l’équipe caractérise comme 'SLA précoce'.
Heureusement, il serait possible d’éviter que cela ne se produise, à partir du moment où les chercheurs arrivent à désactiver le gène mutant SPTLC1 en utilisant de petits ARN interférents (siRNA): des molécules d’ARN qui visent l’allèle mutant en particulier et qui bloquent la surproduction de la SPT.
Si l’expérience n’a été effectuée à présent que sur des cellules de patients – et pas encore sur les patients eux-mêmes – les progrès balisent un avenir permettant de traiter la condition et d’épargner aux enfants la pire des maladies débilitantes et désespérantes.
« Ces résultats préliminaires suggèrent que nous serons peut-être capables de mettre en place une stratégie de désactivation de gène précise pour traiter les patients souffrant de ce type de SLA », affirme le chercheur principal du NINDS Carsten Bönnemann.
« Notre but ultime est de transposer ces idées en traitements effectifs pour nos patients qui n’ont actuellement pas d’options thérapeutiques. »
Les résultats sont repris dans Nature Medicine.
Traduction: Vicky Roels
Source: ScienceAlert