

''Inversions SLA'' : caractéristiques démographiques, caractéristiques de la maladie, traitements et comorbidités
10-04-2018
Résumé
Objectif : identifier les différences entre les caractéristiques démographiques, les caractéristiques de la maladie, les traitements et la comorbidité (présence d'un ou de plusieurs troubles associés à une maladie primaire) des patients présentant des '' inversions SLA'' et ceux atteints de SLA plus généralement progressive.
Méthodes : des cas possibles d’inversion SLA ont été trouvés dans des publications antérieures, à la clinique Duke SLA, dans des références d’autres neurologues et sur l’internet. De 89 inversions potentielles identifiées, 36 ‘cas’ ont été retenus parce que l’examen de données ou de la littérature a confirmé leur diagnostic et une amélioration significative et soutenue dans au moins une mesure objective. Les patients retenus étaient repris dans la ‘Pooled Resource Open-Access ALS Clinical Trials database’ et le ‘National ALS Registry’. Les ‘cas’ et les ‘témoins’ (la population- contrôle) ont été comparés à l’aide de statistiques descriptives.
Résultats : les ‘cas’ d’inversion SLA étaient plus susceptibles d’être de sexe masculin, souffrant d’une SLA qui s’est d’abord développée dans les membres et a, initialement, progressé plus rapidement. Les prévalences de la myasthénie (faiblesse musculaire) grave (MG) et d’affections des motoneurones inférieurs étaient plus élevées chez ces ‘cas’ que celles des estimations des taux de prévalence dans la population générale. Les consommations de curcumine, de lutéoline, du cannabidiol, de l’azathioprine, de cuivre, de glutathion, de vitamine D et d’huile de poisson étaient plus importantes que celles des ‘témoins’.
Conclusions : comparativement aux patients atteints de SLA plus généralement progressive, les patients SLA présentant des inversions diffèrent par leurs caractéristiques démographiques, les caractéristiques de leur maladie et leurs traitements. Si certains de ces patients pourraient souffrir d’une maladie d’anticorps rare mimique de la SLA, comme la myasthénie atypique, les détails de leurs examens, leurs résultats d’électromyographies (EMG) et leurs histoires familiales font valoir que cela est peu probable. Nos résultats suggèrent plutôt que les inversions SLA justifient l’évaluation des mécanismes de résistance à la maladie et que les traitements associés à des inversions SLA méritent une étude plus approfondie.
Introduction
La Sclérose latérale amyotrophique (SLA) est une maladie neurodégénérative dévastatrice et presque toujours fatale. Il est très rare que la SLA cesse de progresser et que le patient regagne une fonction motrice importante. Étudier ces cas d’ inversions SLA pourrait aider à découvrir un syndrome mimique de la SLA sous-diagnostiqué, un mécanisme génétique de résistance à la SLA ou, éventuellement, un traitement SLA efficace. Ici, nous compilons des cas vérifiés d’inversions SLA dans une base de données pour comparer leurs caractéristiques démographiques, leurs caractéristiques de la maladie, leurs traitements et leur comorbidité à ceux des patients atteints de SLA plus généralement progressive.
Méthodes
Sources des données et méthodologie
Il s’agit d’une étude de ‘cas’-‘témoins’. Les cas potentiels ont été identifiés dans des publications antérieures examinées par des pairs (n = 23), de la clinique de Duke ALS (n = 3), par d’autres neurologues (n = 40) et des témoignages postés sur l’internet (n = 23). Les patients chez qui nous avons pu confirmer indépendamment un diagnostic SLA ou d’atrophie musculaire progressive (AMP) et une amélioration significative et soutenue dans au moins une mesure objective par le biais de révision soit des données soit de la littérature consacrée aux inversions SLA ont été retenus comme ‘cas’. Les ‘cas’ ont été diagnostiqués SLA selon critères ‘ El Escorial-Revised’ et/ou critères d’Awaji. Les inversions étaient le plus souvent mesurées par la résolution d’une dénervation sur l’électromyogramme (EMG), des tests de résistance musculaire manuelle et/ou des gains d’au moins 4 points sur l’échelle de mesure fonctionnelle SLA (ALSFRS-R). Dans d’autres cas, des patients présentant des améliorations extraordinaires dans les activités quotidiennes ont également été inclus. Par exemple, nous avons inclus un ‘cas’ qui, à un stade, était incapable de se tenir debout, mais qui, après son inversion, était capable de marcher plusieurs kilomètres. Il n’avait cependant pas subit de test de force musculaire formellement documenté. Les patients présentant une rechute, de retour ou inférieure à leur stade précédent, ont été exclus.
Plus de détails concernant la démographie, les diagnostics, les inversions, les traitements et les comorbidités des cas ont été repris dans une base de données. La population de contrôles (n = 10 723) étaient les patients repris dans la base de données ‘Pooled Resource Open-Access ALS Clinical Trials (PRO-ACT) database’, téléchargée le 17 octobre 2016, transmise par les membres du ‘Pro-ACT Consortium’ (Prize4Life, Northeast ALS Consortium et ALS Therapy Alliance). Les données d’histoire démographique et familiale étaient disponibles pour la comparaison à partir du portail en ligne du ‘National ALS Registry‘ (n = 6 352).
Analyse statistique
Les analyses statistiques ont été réalisées à l’aide de JMP® Pro 13.0.0. Khi et deux échantillons tests-t ont été utilisés pour comparer les caractéristiques démographiques et les caractéristiques des maladies de la population de contrôle. L’évaluation de progression du score de marche a été effectuée avec une analyse de covariance. Des tests de Wilcoxon (rank sum) ont été utilisés pour comparer les tranches d’âge. Les traitements ont été comparés à l’aide de la régression logistique avec correction de Bonferroni.
Approbations des standards du protocole, inscriptions et consentements des patients
Cette étude a été approuvée par la Commission institutionnelle de la Duke University. Il n’y a aucune intervention médicale ou information privée (protégée) de santé enregistrée au cours de cette étude. En raison du très faible risque, une dispense de consentement a été obtenue.
Résultats
Diagnostics et inversions
Un total de 36 ‘cas’, définis cliniquement SLA(n = 4), cliniquement probables SLA ou cliniquement probables SLA après test en laboratoire (n = 23), possibles SLA sur le plan clinique (n = 2) et d’atrophie musculaire progressive (n = 7) ont été identifiés à travers la littérature (n = 16) et l’examens de dossiers(n = 20). Les informations sur leurs diagnostics et leurs inversions sont inclues dans les tableaux complémentaires 1 et 2. Tous avaient un historique, subi des examens neurologiques, des électromyogrammes (EMG) et de tests pour des mimiques SLA, ce qui rends la SLA encore plus probable. À noter que 19 % des cas avaient un syndrome purement de motoneurones inférieurs par rapport à une estimation de 5 % de la population de tous les patients atteints de maladies du motoneurone. Nous avons considéré ces patients ''d’atrophie musculaire progressive'' comme atteints de la SLA pour les raisons exposées précédemment.
Après une baisse initiale, le délai médian du maximum de l’amélioration a été 12 mois (intervalle 1– 206 mois). 16 ‘cas’ s’étaient améliorés jusqu’à leur dernière visite de suivi connue. Pour les 18 ‘cas’ qui avaient atteint un plateau après leur amélioration maximale, la durée médiane du suivi était de 38,5 mois (rang 3 – 295 mois). La durée de suivi n’a pas pu être déterminée dans 2 ‘cas’ (participants 24 et 26), leurs dates d’amélioration maximale n’étant pas disponibles. Les mesures utilisées pour suivre la progression de la maladie varient d’un ‘cas’ à l’autre selon la disponibilité dans le dossier ou le rapport. Les cas potentiels avec des améliorations sur une ou plusieurs mesures ont été exclus s’ils montraient des détériorations sur toutes les autres mesures. Il y avait 12 ‘cas’ avec des améliorations sur le ALSFRS-R (moyenne de 9,6, SD 4,7 points). Les progressions des scores ALSFRS-R de l’apparition de la maladie à la dernière visite de suivi connue sont reprises dans la Figure 1. Lors des améliorations en force sur le test musculaire manuel, trois ou quatre membres se sont améliorés dans la majorité des ‘cas’. Il y avait 9 ‘cas’ revenus à une force normale et 7 ‘cas’ avec pleine résolution de la dénervation active sur EMG au moment de leur inversion.
Démographie et caractéristiques de la maladie
Des comparaisons entre la démographie et les caractéristiques des maladies des ‘cas’ et des ‘témoins’ sont inclus dans le tableau 1. Le taux de progression de la maladie a été mesuré par la perte de points/an sur le score de marche ALSFRS-R parce que seulement quelques ‘cas’ disposaient des scores ALSFRS-R en entier au moment de leurs nadirs. Après ajustement selon l’âge et l’emplacement de la déclaration de la maladie, les taux de progression des scores de marche ALSFRS-R des ‘cas’ sont restés significativement plus rapides que ceux des ‘témoins’.
Comorbidités
Ni ‘PRO-ACT’ ni le ‘National ALS Registry’ ne contiennent des informations complètes sur les comorbidités de leurs participants. Toutefois, la prévalence d’un diagnostic préalable de myasthénie grave (MG) dans nos ‘cas’ (6 %, n = 2, les participants 34 et 35) était plus élevé que les estimations de prévalence dans la population générale (0,03 %), même si la SLA et la MG peuvent coexister plus fréquemment que prédit par hasard seul.
Traitements
Le tableau complémentaire 3 contient des informations sur les traitements utilisés par au moins deux ‘cas’ au moment de leur amélioration maximale et pour lesquels les occurrences de prendre ce traitement étaient significativement plus élevées que pour les ‘témoins’ de ‘PRO-ACT’. Tous ces traitements, à l’exception de l’azathioprine, ont été utilisés exclusivement par des ‘cas’ recensés à travers l’examen de dossiers. Après ajustement selon l’âge et l’emplacement d’apparition de la maladie, les occurrences de prise de curcumine, d’azathioprine, de cuivre, de glutathion, de vitamine D et d’huile de poisson étaient significativement plus élevées chez les ‘cas’ que chez les ‘témoins’. Des données insuffisantes empêchaient ces ajustement chez les patients qui ont utilisé la lutéoline ou le cannabidiol, mais les occurrences de prise de ces deux traitements sont restées plus élevées chez les ‘cas’ que chez les ‘témoins’ après ajustement selon l’âge à l’apparition de la maladie.
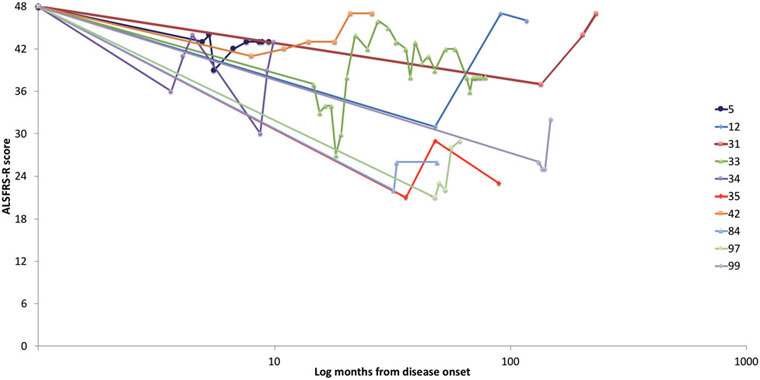
Figure 1 : la progression ALSFRS-R. Il y avait 12 ‘cas’ avec des améliorations mesurées par le ALSFRS-R. Parmi eux, 10 sont inclus dans la figure (le ‘cas’ 38 a eu des mesures initiales ALSFRS avant l’introduction de l’ALSFRS-R et le ‘cas’ 24 a une date inconnue d’amélioration maximale). Ce graphique montre l’évolution au fil du temps de ces 10 cas telle que mesurée par le ALSFRS-R de l’apparition de la maladie à la dernière visite de suivi connu. Les participants 5, 33, 34, 35, 42 et 99 avaient documenté leurs améliorations sur d’autres mesures objectives additionnelles. ALSFRS(-R) : mesure fonctionnelle SLA (-révisée).
Tableau 1. Démographie et caractéristiques des maladies des inversions SLA.
| A | Cas | PRO-ACT | Test statistique | Résultat |
| Age apparition de la SLA | 50.1 (15.3) | 53.8 (11.8) | t = 1.89 | p = 0.0588 |
| % Hommes | 80.6 | 60.3 | χ2 = 6.14 | p = 0.0132 |
| % Blancs | 87.5 | 95.4 | χ2 = 3.36 | p = 0.0669 |
| % Antécédents familiaux SLA | 8.00 | 15.7 | χ2 = 1.10 | p = 0.2940 |
| % SLA membres | 93.9 | 72.1 | χ2 = 7.80 | p = 0.0052 |
| Taux progression ‘de marche’ | −1.59 (1.81) | −0.991 (0.762) | t = 4.28 | p < 0.0001 |
| B | Cas | National ALS Registry | Test statistique | Résultat |
| Age du diagnostic | 50–59 | 50–59 | Z = 2.88 | p = 0.0040 |
| % Hommes | 80.6 | 60.5 | χ2 = 6.05 | p = 0.0139 |
| % Blancs | 87.5 | 95.2 | χ2 = 3.08 | p = 0.0792 |
| % Antécédents familiaux SLA | 4.00 | 4.20 | χ2 = 0.003 | p = 0.9594 |
ALS: amyotrophic lateral sclerosis; ALSFRS-R: ALS Functional Rating Score–Revised; PRO-ACT: Pooled Resource Open-Access ALS Clinical Trials.
Ce tableau compare les données démographiques disponibles et les caractéristiques de la maladie des patients atteints de SLA et présentant des inversions aux patients repris dans la base de données de ‘Pro-ACT’ (A) et du ‘National ALS Registry’ (B). Les antécédents familiaux dans (A) incluent les parentés du premier au troisième degré et les données sur les antécédents familiaux dans (B) incluent les ascendants du premier degré. À ce titre, étaient considérés comme des cas SLA avec antécédents familiaux SLA ceux dans (A) dont des parents jusqu’au troisième degré étaient atteints de SLA et dans (B) ceux dont des parents du premier degré étaient atteints de SLA. Les tranches d’âge étaient seulement disponibles (B), les patients ayant été comparés à l’aide de tests de Wilcoxon. Les tranches d’âge médian étaient équivalentes, cependant, la somme des tranches d’âge des ‘cas’ était significativement moins élevée et la somme des tranches d’âge des ‘témoins’ significativement plus élevée que leurs valeurs attendues. Les proportions ont été analysées avec les tests khi-carré de Pearson et les moyennes avec deux échantillons tests-t. Les écarts-types sont repris, le cas échéant, entre parenthèses. Le taux de progression du score de marche est mesuré en tant que changement annuel en points sur la question ‘ marche’ de ALSFRS-R.
Commentaires
Pour la première fois, nous avons recensé des cas vérifiés d’inversion SLA dans une base de comparaison aux patients atteints de SLA plus généralement progressive. Il existe certaines différences dans la démographie et les caractéristiques des maladies des ‘cas’ par rapport aux ‘témoins’. Ces différences persistent avec les deux groupes de contrôles et sont compatibles avec les données publiées antérieurement sur les inversions SLA. Il est important de noter que la définition d’inversion SLA utilisée dans cette étude diffère de définitions précédentes: la hausse de minimum quatre points sur le ALSFRS-R sur une durée minimale de douze mois. Notre nouvelle définition permet d’inclure des patients avec des améliorations significatives qui n’ont pas de scores ALSFRS-R. Par exemple, un de nos ‘cas’ sans score ALSFRS-R est sorti de l’assistance respiratoire après 17 années de dépendance et un autre a recommencé à marcher après une année de quadriplégie. Notre nouvelle définition exige également que les améliorations soient robustes et durables, parce que de petites améliorations transitoires en ALSFRS-R et dans la force musculaire ne sont pas rares chez les patients atteints de SLA généralement progressive. Des préoccupations existaient concernant des travaux antérieurs sur les inversions SLA suite à des difficultés d’interprétations de la base de données ‘PRO-ACT’ et l’inclusion d’améliorations de résultats ALSFRS-R sans la confirmation par d’autres données de mesures et le manque d’informations confirmant l’endurance pour les maladies mimiques SLA. La majorité des patients décrits ici avec des améliorations de résultats ALSFRS-R présentait également des améliorations lors de l’examen de la force musculaires ou d’autres mesures. En outre, tous les cas avaient des informations disponibles au sujet de l’endurance en cas de maladie mimique SLA. Si la plupart des patients disposaient d’une vaste imagerie, EMG ou d’études sur la conductivité neuronale/ et des tests d’endurance en laboratoire, ceux-ci variaient largement d’un ‘cas’ à l’autre.
Une des hypothèses possibles est que certains de nos ‘cas’ aient été diagnostiqués à tort de la SLA, souffrant en fait d’une maladie d’anticorps rare mimique de la SLA. Le taux élevé de comorbidité MG et l’occurrence élevée d’utilisation d’azathioprine chez nos ‘cas’ pourrait conforter cette idée. Le ‘cas’ 34, par exemple, avait été diagnostiqué séropositif MG avec réactions d’immunomodulations. Il a été diagnostiqué de la SLA seulement 2 ans plus tard après le développement de réflexes plantaires montants, d’affections pseudobulbaires et des réflexes tendineux vifs dans les bras et les jambes avec des signes de dénervation et de réinnervation sur EMG, en l’absence de décrémentation de la stimulation nerveuse répétitive. Le ‘cas’ 35 a été diagnostiqué avec MG après un épisode subaiguë de ptose, une dysarthrie, une dysphagie et une fatigue qui disparut complètement après 2 mois. Il a été diagnostiqué atrophie musculaire progressive (AMP) 30 – 40 ans plus tard sur base d’atrophie généralisée, de faiblesse (plus distalement dans les extrémités supérieures) et de signes de dénervation et de réinnervation sur l’EMG. Cependant, la MG et la SLA peuvent cohabitent plus souvent que prévu par le hasard seul. L’absence de fluctuations de la faiblesse ou de faiblesse oculaire, la présence de signes de motoneurones supérieurs et la présence d’une dénervation EMG dans la plupart de nos cas sont des arguments forts pour décider que la MG seule n’explique pas tout. Le taux similaire d’antécédents familiaux SLA chez nos ‘cas’ et chez les ‘témoins’ écarte encore davantage l’idée que certains de nos ‘cas’ aient été mal diagnostiqués.
Les occurrences de prise de curcumine, de lutéoline, de cannabidiol, d’azathioprine, de cuivre, du glutathion, de vitamine D et d’huile de poisson sont plus élevées chez les ‘cas’ que chez les ‘témoins’. Il est possible qu’un biais de déclaration ou de sélection existe concernant la prise du cannabidiol chez les ‘témoins’, la toxicomanie excluant de participer à certains essais inclus dans Pro-ACT. L’association ne prouve pas le lien de causalité. Cependant, les huit thérapies identifiées ici sont particulièrement intéressantes parce qu’elles ont des mécanismes pouvant plausiblement influencer la SLA. Par exemple, la lutéoline, le glutathion et la vitamine D atténuent le stress oxydatif et l’huile de poisson, le cannabidiol et l’azathioprine réduisent l’inflammation. En outre, chaque thérapie identifiée ici a été temporairement associée à au moins deux inversions. Ces thérapies devraient encore être évaluées dans des études prospectives.
Enfin, il est possible que nos ‘cas’ présentent des différences génétiques qui confèrent une résistance à la maladie. Il existe un précédent pour cela dans les minorités contrôlant le VIH. Cette découverte a conduit à un traitement efficace pour les patients atteints du VIH, le Maraviroc. Il est possible que certains de ces ‘cas’ aient des mutations qui mènent à une réinnervation améliorée. Cela pourrait être particulièrement bénéfique dans les cas de maladies du motoneurone inférieur, présentes parmi nos ‘cas’. Nous prévoyons de réaliser le séquençage du génome entier des ‘cas’ d’inversions SLA pour déterminer s’ils diffèrent génétiquement de patients avec une progression SLA plus typique. En comprenant mieux les inversions SLA, nous espérons les faire se produire plus souvent.
Remerciements
Les auteurs sont reconnaissants à tous les patients SLA et à leurs familles, qui ont partagé leurs témoignages dans l’espoir qu’ils pourraient aider d’autres pALS. Sans leur temps et leur intérêt, cette étude n’aurait pas été possible. Nous remercions également le Dr Edwin Iversen pour son aide et le ‘Duke University Statistical Consulting Center’ pour leurs soutiens en méthodes statistiques. Ce travail a été soutenu par la ‘LVH ALS Foundation’ [licence numéro 1].
Traduction : Fabien
Source : Taylor & Francis