

Pour lutter contre les maladies du cerveau, les médicaments visent l’accumulation de trafic protéique à l'intérieur de la cellule, qui tue les neurones
21-01-2019

L'oeil de mouche composé normal (à gauche) est gâché par la mort cellulaire dans une souche (à droite) portant une mutation causant la sclérose latérale amyotrophique.
Les yeux composés de la mouche commune des fruits sont normalement rouge brique. Mais dans le laboratoire du neurologue Tom Lloyd de la faculté de médecine de l'Université Johns Hopkins à Baltimore dans le Maryland, de nombreux yeux de mouche sont couverts de taches blanches et noires, signe que les neurones de chacune de leurs quelque 800 unités oculaires sont en train de se ratatiner et de mourir.
Ces mouches ont l'équivalent génétique de la sclérose latérale amyotrophique (SLA), le trouble neurodégénératif débilitant également connu sous le nom de maladie de Lou Gehrig, et leurs yeux offrent une fenêtre sur le processus de la maladie. En mesurant l'étendue des dommages aux yeux de chaque insecte, les chercheurs peuvent rapidement déterminer si un médicament, une modification génétique ou une autre intervention thérapeutique aide à enrayer la perte neuronale.
Ces yeux ont également apporté une réponse au mystère central de la SLA: ce qui tue les neurones et, en fin de compte, le patient.
Les mouches portent une mutation retrouvée chez environ 40% des patients SLA ayant des antécédents familiaux de la maladie et chez environ 10% des cas sporadiques. La mutation, dans un gène appelé C9orf72, consiste en des centaines ou des milliers de copies supplémentaires d'une courte séquence d'ADN de six bases seulement. Ils entraînent la formation de brins d'ARN d'une taille inhabituelle qui recouvrent des centaines de protéines dans le noyau de la cellule, ce qui les met hors de combat. Lloyd et son collègue de Hopkins, Jeffrey Rothstein, ont émis l'hypothèse que certaines de ces protéines contenues dans l'ARN pourraient être la clé de la SLA.
Pendant plusieurs mois, les chercheurs ont étudié de manière systématique le rôle de chaque protéine en développant des souches de mouches portant à la fois la mutation de la SLA et une version avec incapacité ou hyperactivité du gène de chaque protéine. Un groupe de mouches, élevé pour avoir des niveaux élevés d'une protéine appelée RanGAP, s'est démarqué. Quinze jours après que les mouches eurent émergé de leur enveloppe nymphale, leurs yeux restèrent une pure terre de Sienne brûlée. RanGAP "était de loin le suppresseur le plus puissant de la neurodégénérescence", déclare Lloyd. Ce qui était connu à propos de sa fonction était tentant: il sert de courrier, aidant à faire passer d’autres protéines à travers la membrane qui sépare le noyau cellulaire du cytoplasme.
Les résultats de l'équipe permettraient aux neuro-scientifiques de mieux comprendre la SLA et les maladies du cerveau en général, tandis que d'autres étaient sur la même piste. En 2015, deux autres équipes de recherche ont indiqué que les défauts du système de transport nucléaire de la cellule étaient des caractéristiques non seulement de la SLA, mais également de la démence frontotemporale (DFT), une autre maladie cérébrale évolutive causée par des mutations C9orf72. Les scientifiques associeraient bientôt le trafic dysfonctionnel de la fracture nucléaire à d’autres maladies neurodégénératives (Alzheimer, Huntington, ataxie spinocérébelleuse) et même au vieillissement normal. Dans tous ces maux, les entassements anormaux de protéines qui en résultent deviennent en quelque sorte des tueurs neuronaux indésirables.
"J'ai souvent mal à la tête lorsque quelqu'un fait une découverte et essaie de l’expliquer au reste du monde", a déclaré Rothstein, neurologue à la tête du Johns Hopkins Brain Science Institute. Mais ici, dit-il, cela semble être vrai.
Les résultats ne sont pas simplement académiques. Ils inspirent les efforts thérapeutiques pour s'attaquer à la cause de la neurodégénérescence liée à l'âge - un objectif qui a largement échappé aux développeurs de médicaments. Si la perte progressive du transport nucléocytoplasmique est une caractéristique conservée du cerveau vieillissant, explique Sami Barmada, neurologue à l'Université du Michigan à Ann Arbor, sa prévention "pourrait constituer un traitement vraiment vaste et efficace".
Plusieurs sociétés de biotechnologie ont sauté sur cette idée, l'explorant dans des modèles animaux et planifiant les premiers essais sur l'homme cette année. Parmi eux: Biogen à Cambridge, dans le Massachusetts, qui a acheté en 2018 les droits pour développer un composé pharmaceutique appelé KPT-350 qui cible directement la voie de transport nucléaire. La recherche qui sous-tend l'action de ce médicament est nouvelle. Mais "La biologie est là", déclare Chris Henderson, responsable de la recherche sur les troubles neuromusculaires et du mouvement chez Biogen. "Voici un médicament avec une logique d’existence", ajoute-t-il, "et nous sommes optimistes quant à l'inclusion de cette substance dans les essais."
Traverser la frontière nucléaire
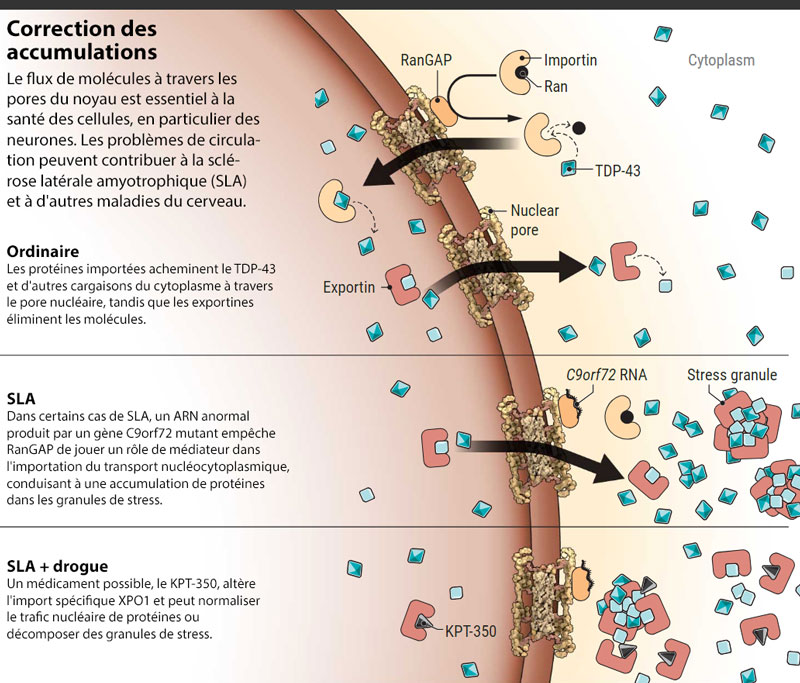
La membrane lipidique qui sépare le noyau riche en ADN du reste de la cellule est comme une frontière internationale occupée par un trafic industriel à double sens. Des protéines de liaison à l'ADN et d'autres molécules affluent constamment dans le noyau pour aider à activer et désactiver les gènes, par exemple. Les ARN messagers produits par ces gènes s'écoulent dans le cytoplasme jusqu'aux plateformes d'assemblage de protéines. La cellule doit réguler ce trafic par des points d’entrée appelés pores nucléaires. Étouffer ces portails et il va de soi que les cellules vont en souffrir.
La première idée qui pourrait perturber le transport nucléaire pourrait sous-tendre la SLA est apparue en 2010, lorsque des chercheurs du King's College de Londres, travaillant avec des cellules cancéreuses nerveuses, ont bloqué de manière expérimentale l'expression des protéines impliquées dans le transport d'importation. Le résultat a également été observé dans les cellules de patients atteints de SLA: des amas d'une protéine appelée TDP-43 s'accumulant dans le cytoplasme.
Peu de chercheurs de la SLA ont prêté une grande attention à ce premier rapport. Ce qui pourrait gommer les rouages de la machinerie de transport chez les patients atteints de SLA n'était pas clair, et les chercheurs ne pouvaient pas dire si l'accumulation de TDP-43 - une protéine qui lie normalement à la fois l'ADN et l'ARN à l'intérieur du noyau pour réguler plusieurs étapes dans l’expression des gènes - était en train de tuer des neurones ou était simplement la conséquence d’un processus toxique différent. Il faudrait encore cinq ans - et l'étude de Lloyd's et Rothstein sur les mouches d'un œil révélateur - pour que les scientifiques de la SLA prennent les échanges entre le noyau et le cytoplasme dans les deux sens (importation et exportation) plus au sérieux.
“C’est un médicament qui a une logique, et nous sommes optimistes quant à son inclusion dans les essais.”
Chris Henderson, Biogen
Les résultats de l’équipe Hopkins ont électrisé les collègues en partie parce qu’ils avaient identifié une protéine de transport, RanGAP, comme facteur clé de la neurodégénérescence. L'équipe a montré à la fois dans le modèle de mouche de la SLA et dans des cellules de patients humains que les longues lectures d'ARN produites par le gène C9orf72 mutant semblaient se coller à RanGAP près du pore nucléaire et mettre la protéine hors d'usage. La perte de fonctionnement de RanGAP a entraîné une sauvegarde du système d'importation nucléaire, entraînant une accumulation cytoplasmique de protéines telles que TDP-43, encombrant une cellule ressemblant à des sacs de déchets pourris lors d'une frappe-poubelle.
L'équipe a été tout aussi convaincante de constater qu'un médicament potentiel pourrait préserver la santé neuronale, du moins chez les mouches. "Tout à coup, un traitement potentiel a été lancé sur le ring", déclare Dorothee Dormann, biochimiste à l'Université Ludwig Maximilian de Munich, en Allemagne.
L'équipe ne disposait d'aucun médicament susceptible d'augmenter les niveaux de RanGAP dans le cytoplasme et de restaurer suffisamment de flux entrant pour sauver les neurones de l'œil. Mais Lloyd a estimé que bloquer le flux sortant de TDP-43 et d'autres protéines nucléaires pourrait avoir le même effet bénéfique. Un composé expérimental appelé KPT-276 était connu pour inhiber sélectivement une protéine clé d'exportation nucléaire appelée exportine 1 (XPO1). L'approche consistait en une sorte de piratage, associant deux erreurs - une entrée et une sortie défectueuses - pour créer une droite, mais cela a fonctionné. Lorsque Lloyd a donné le KPT-276 à ses mouches SLA, leurs yeux sont restés vierges.
Du combattant du cancer au protecteur du cerveau
KPT est le code de composé expérimental utilisé par Karyopharm Therapeutics, une petite société pharmaceutique basée à Newton, dans le Massachusetts. Karyopharm a été créée en 2008 pour développer des inhibiteurs de XPO1 destinés au traitement du cancer, l’idée étant de déclencher une accumulation de protéines suppresseurs de tumeurs dans le noyau, où ils exercent leur fonction de chien de garde anticancéreux.
Une décennie plus tard, le premier candidat clinique de la société, un médicament contre le myélome multiple, devrait largement obtenir l’autorisation de mise sur le marché dans les mois à venir.
Les chimistes de Karyopharm ont mis au point une série d'inhibiteurs de la XPO1, notamment le KPT-276 et un parent appelé KPT-350, qui présentaient un attribut important: ils franchissaient la barrière hémato-encéphalique plus facilement que les autres candidats. Le KPT-350 s'est avéré plus puissant et moins toxique lors des tests précliniques. L'entreprise a donc cherché des moyens de l'utiliser pour traiter les maladies et les lésions cérébrales.
Les résultats de Lloyd's et Rothstein ont piqué l'intérêt de la société. Lorsque Sharon Tamir, responsable des maladies neurodégénératives et infectieuses à l'époque, a appris que les chercheurs de Hopkins travaillaient avec KPT-276 et non pas KPT-350, elle les a appelés à proposer une collaboration utilisant le "meilleur" composé. Parallèlement, elle a commencé à distribuer le KPT-350 à d’autres groupes au Japon, en Belgique et aux États-Unis. Ensemble, ces scientifiques ont montré les effets neuroprotecteurs du médicament sur toute une gamme de modèles de cellules SLA, de mouches et de rongeurs humains, de maladie de Huntington et d'autres maladies du cerveau.

Une micrographie à force atomique en fausse couleur montre les pores complexes (anneaux verts) qui régulent étroitement le trafic dans la cellule entre le noyau et le cytoplasme. - Source: H.oberleithner, University Hospital Of Muenster/science
Une équipe dirigée par le neuroscientifique Jeffery Haines à Icahn. De 'école de médecine du mont Sinaï à New York a démontré, par exemple, que le traitement au KPT-350 a préservé la santé des axones, les longues extensions des cellules nerveuses transmettant le signal, et amélioré les fonctions motrices de souris présentant une maladie semblable à la sclérose en plaques. Et dans les labos du groupe Hopkins, le médicament a gardé en vie les neurones de souris hébergeant la mutation associée à Huntington.
"Il reste encore beaucoup à faire pour expliquer pourquoi le complexe de pores nucléaires est si vulnérable aux problèmes de différents types de neurones dans différentes régions du cerveau, responsables de multiples maladies différentes", a déclaré Gavin Daigle, ancien postdoc du laboratoire de Rothstein, qui a travaillé sur le projet. Avant de rejoindre AbbVie à Cambridge, le projet Huntington a permis de relier la perturbation de la fonction des pores à la maladie d’Alzheimer. Mais il souligne que toutes les recherches montrent une chose: "C’est une voie qui peut être ciblée".
Les résultats ont suffi à convaincre Biogen, qui a acheté les droits pour tester le médicament chez l'homme. "L'ensemble des données précliniques que Karyopharm a pu rassembler justifie réellement l'excitation", déclare Laura Fanning, chef de projet R & D pour KPT-350 chez Biogen (qui a renommé la molécule BIIB100). "Ce n'est pas juste un coup d'efficacité sur une souche de souris. C'est une vaste base de preuves", dit-elle. Une première étude d’augmentation de la dose de KPT-350 chez l’homme pourrait commencer chez les patients SLA plus tard cette année. Si le médicament semble prometteur contre cette maladie, Biogen pourrait étendre ses tests cliniques à d’autres maladies, dit Henderson.
En route pour les essais cliniques
Bien que le médicament semble fonctionner en laboratoire, le pourquoi ou comment ne sont pas clair du tout. "L'histoire a commencé à devenir plus sombre à mesure que davantage de données sont entrées", note Haines, maintenant à Regeneron Pharmaceuticals à Tarrytown, dans l'État de New York. Au départ, la plupart des scientifiques ont supposé que, du fait de son blocage du XPO1, le médicament empêchait des protéines telles que TDP-43 de s'accumuler dans le cytoplasme en les emprisonnant dans le noyau. Mais l'année dernière, l'équipe de Dormann et une autre dirigée par Philip Thomas, biochimiste du Southwestern Medical Center de l'Université du Texas à Dallas, ont rapporté de manière indépendante que TDP-43 et une autre protéine appelée FUS semblaient sortir du noyau par diffusion passive et non par XPO1-. transport médiatisé. (Les FUS s'agglutinent également dans le cytoplasme des motoneurones chez certains patients atteints de SLA ou de DTF.)
Donc, si le KPT-350 n'agit pas directement sur le système de transport, que fait-il? "Il semble que le médicament cible une voie neurotoxique plus générale", a déclaré Dormann, "mais il reste à préciser le mécanisme réel et les défauts de transport nucléaire que nous corrigeons avec ce médicament."
Une recherche récente suggère que l'une des possibilités est que le médicament cible réellement de minuscules paquets denses de protéines et d'ARN qui se forment pendant les périodes de stress cellulaire. Dans les cellules saines, ces "granules de stress" sans membrane se décomposent généralement et leurs composants se dispersent après le passage d'une infection virale, d'un choc thermique ou d'une autre agression de l'environnement. Ce n’est pas le cas dans les neurones malades des personnes atteintes de SLA ou de DTF. Dans ces cellules, les granules de stress persistent et deviennent collantes, recrutant des protéines telles que TDP-43 et FUS et finissant par se transformer en agrégats solides et toxiques.
Au cours de la dernière année, plusieurs équipes de recherche ont montré que des composants des équipements de transport nucléaire - y compris des importateurs, des exportateurs et des parties du pore nucléaire lui-même - pouvaient également être emmêlés dans ces agrégats. Le système de transport faiblit et, alors que de plus en plus de protéines TDP-43 et d’autres protéines s’ajoutent aux granules de stress, une boucle de rétroaction s’installe qui bloque le trafic moléculaire. "Le TDP-43 n'est pas seulement une victime de défauts de transport nucléo-cytoplasmique", explique Wilfried Rossoll, neuroscientifique à la Mayo Clinic de Jacksonville, en Floride. "C'est aussi un auteur"
En août 2018, les résultats d'une étude menée par le neurobiologiste Ludo Van Den Bosch de l'Université catholique VIB de Louvain en Belgique suggèrent que la protéine de transport XPO1 pourrait elle-même jouer un rôle dans les granules de stress. Cela signifie qu'un médicament tel que le KPT-350 peut principalement servir d'agent anti-stress, et que tout impact sur le transport peut être secondaire. "Les choses sont plus compliquées qu'initialement présentées", déclare Van Den Bosch, qui a collaboré avec Karyopharm.
Les questions en suspens sur KPT-350 n'ont pas découragé les autres groupes de poursuivre des stratégies supplémentaires pour résoudre les problèmes de trafic nucléaire. En 2017, par exemple, Guillaume Hautbergue et ses collègues de l'Université de Sheffield au Royaume-Uni ont impliqué un autre facteur d'exportation dans la perte neuronale subie par les mouches de la SLA portant la mutation C9orf72. Hautbergue travaille sur des moyens de cibler cette protéine afin d'empêcher l'exportation d'ARN mutants produits par le gène.
D'autres chercheurs se concentrent sur la fragmentation des granules de stress. Cette approche devrait libérer les facteurs de transport et les protéines des pores retenus en otage dans ces granules, leur permettant ainsi de reprendre leurs fonctions habituelles dans la cellule, explique James Shorter, biochimiste des protéines à l'Université de Pennsylvanie.
Il développe un moyen d'équiper les cellules avec un gène pour la fabrication d'une protéine "désagrégase" et a commencé à tester la stratégie thérapeutique dans un modèle souris de SLA.
Quelques sociétés pharmaceutiques, dont Denali Therapeutics de South San Francisco, Californie, et Aquinnah Pharmaceuticals de Cambridge, recherchent des petites molécules capables de faire la même chose. Ces thérapies ne cibleront peut-être pas directement la voie du transport nucléaire, mais elles accompliraient leur travail, explique Ben Wolozin, cofondateur et directeur scientifique d'Aquinnah, neuropharmacologue à la School of Medicine de l'Université de Boston, car le démantèlement de granules de stress contribue à rétablir le transport nucléaire.
"Tout cela fait partie d'une réponse biologique intégrée", a déclaré Wolozin.
Aquinnah espère pouvoir commencer à évaluer son composé principal chez les patients atteints de SLA cette année, à peu près au même moment où Biogen vise à introduire le KPT-350 au niveau clinique. Pour le moment, les scientifiques de Biogen tentent toujours de cerner ce que le médicament fait dans divers modèles génétiques de la maladie, y compris les mouches aux yeux brouillés.
Henderson ajoute que dans une certaine mesure, connaître le mécanisme d'action exact n'a pas vraiment d'importance. "L'expérience pertinente", conclut-il, "est chez le patient humain".
Traduction : Christina Lambrecht
Source : Science