

Une nouvelle thérapie génique potentielle pour la SLA franchit une étape-clé
23-05-2017
News Analysis, Michelle Pflumm
Environ 1 sur 5 cas de SLA familiale se produit en raison de mutations SOD1 (gène codant l’enzyme superoxyde dismutase-1). La façon dont ces changements génétiques contribuent à la SLA reste inconnue. Mais, selon un nombre croissant d’études, ces mutations entraînent le mauvais repliement et l’agrégation de cette enzyme métabolique, ce qui contribue à la toxicité des motoneurones par plusieurs mécanismes (voir Taylor et al., 2016).
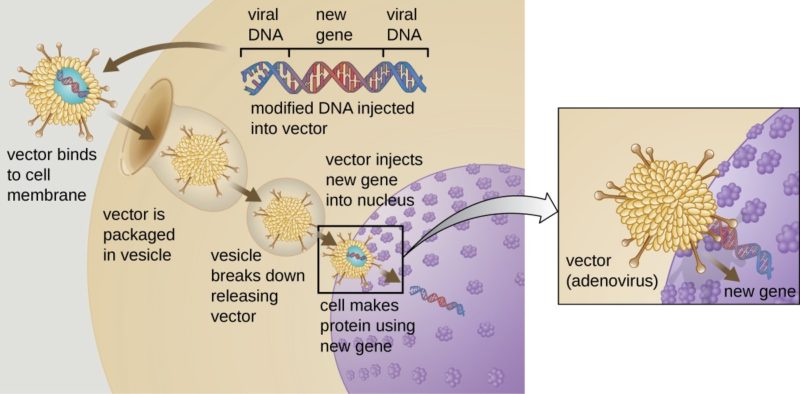
Votre attention, s’il vous plaît. Des scientifiques développent des stratégies de thérapie génique pour la SLA SOD1 qui visent à réduire le nombre de SOD1 mutants dans les principaux tissus touchés par la maladie, y compris le cerveau et la moelle épinière. [Image : Rice University under a CC BY 4.0 license]
Un nombre croissant de chercheurs mettent au point des stratégies de traitement potentiels visant à réduire les niveaux de SOD1 mutants dans le SNC (Système Nerveux Central), dans l’espoir de ralentir ou d’arrêter la progression de la maladie. Une des approches, une thérapie génique, consiste à supprimer l’expression du gène SOD1 par des mécanismes de silençage ARN. Cette stratégie, selon une étude dirigée en 2016 par le Dr Christian Mueller du Medical School of the University of Massachusetts, peut prolonger jusqu'à 20 % la durée de vie de souris adultes SOD1 par une injection intrathécale avant apparition (Borel et al., 2016).
Actuellement, les Dr Martine Barkats et Maria Grazia Biferi, de l’Institut de Myologie de Paris, proposent une nouvelle approche de thérapie génique qui augmente la survie de souris adultes présymptomatiques SOD1 G93A de plus de 50 %. Une analyse indépendante, commandée par Prize4Life, et dirigée par le Dr. Fernando Vieira de l’ALS Therapy Development Institute, a confirmé les résultats de l’équipe.
L’équipe de l’Institut de Myologie de Paris a reçu un prix de 1 million USD (Prize4Life $1.0 million USD Avi Kremer ALS Treatment Prize) en reconnaissance de cette étape-clé. Le Dr. Nicole Szlezák, Présidente du Prize4Life’s Board of Directors in the US, a déclaré : « C’est la meilleure efficacité constatée jusqu’à présent chez des souris SOD1 ».
La Dr Maria Grazia Biferi a dévoilé la stratégie du traitement potentiel le 25 avril 2017 à l’ALS Association Drug Company Working Group, qui s’est tenu au cours de la 69e réunion annuelle de l’American Academy of Neurology (AAN 2017).
L’approche s’appuie sur une stratégie développée précédemment par l’équipe du Dr Barkats, une thérapie génique connexe pour l’atrophie spinale musculaire. Une stratégie similaire, connue sous le nom de ChariSMA (AVXS-101), est actuellement testée en phase 1 et elle semble prometteuse selon les résultats intérimaires en phase I (voir news December 2015).
Selon le Dr Lucie Bruijn, directrice scientifique de l’ALS Association (USA) « Ceci est plus qu’un exercice académique. Les essais cliniques actuels pour l’atrophie spinale musculaire confirment que cette approche peut réussir. »
La voie virale
En 2007, l’équipe du Dr Martine Barkats a découvert que le virus recombinant adeno-associé AAV9 pouvait pénétrer la barrière hémato - encéphalique, ouvrant la voie à l’élaboration de thérapies géniques pour les maladies des motoneurones. (voir December 2008 news; Duque et al., 2009; Foust et al., 2009).

A chaque chien, sa voie. Des chercheurs de la ‘Tufts University School of Medicine’ testent actuellement une thérapie génique potentielle pour la SLA SOD1 sur des chiens. L’approche vise à aider des chiens souffrants d’une maladie naturelle, la myélopathie dégénérative canine (DM), maladie à apparition tardive détectée pour la première fois dans les années 1970 chez des bergers allemands. Ces tests sont une étape-clé dans l’élaboration d’un traitement pour la SLA, parce que les chiens sont des animaux plus grands et parce que la maladie est d’origine naturelle. [Image: Handicapped Pets. CC BY-SA 2.0 license.]
S’appuyant sur ces avancées, des équipes de recherche dirigés par le Dr Brian Kaspar du Nationwide Children’s Hospital en Ohio et le Dr Robert Brown du Centre Médical de l’Université du Massachussets, ont commencé à développer des thérapies potentielles pour la SLA SOD1. Une de ces stratégies, développée par le Dr Robert Brown et le Dr Christian Mueller de l’University of Massachusetts Medical Center, utilise un microARN artificiel pour réduire les niveaux de SOD1 mutant dans le cerveau et la moelle épinière. L’approche est en cours d’évaluation au stade préclinique chez des chiens atteints de myélopathie dégénérative (DM), une maladie naturelle. L’essai clinique, dirigé par l‘Assistant Dominik Faissler de la Tufts University School of Medicine dans le Massachusetts, a été lancé en décembre 2016 et est en cours.
Cette stratégie s’appuie en partie sur des études précédentes menées par la Dr Martine Barkats prouvant que l’AAV10 est plus efficace que l’AAV9 pour introduire, à faibles doses, des gènes dans les motoneurones de la moelle épinière, processus critique dans l’élaboration d’un traitement pour une maladie (Tanguy et al., 2015). Ce processus, selon des études menées par le Dr Krystof S. Bankiewicz de l’University of California et le Dr Christian Mueller du San Francisco and University of Massachusetts Medical Center, peut être appliqué efficacement par voie intrathécale dans les motoneurones, au moins chez les primates non-humains.
En même temps, les Prof Ben Deverman et Prof Viviana Gradinaru du California Institute of Technology ont utilisé CREATE (Cre-recombination-based AAV targeted evolution), pour développer de nouveaux vecteurs de livraison de thérapie génique pénétrant la barrière hémato-encéphalique, pour traiter les troubles du système nerveux central, y compris la SLA. Ces vecteurs, dont l’AAV-PHP. B, peuvent fournir des gènes à l’intérieur du cortex moteur cérébral et de la moelle épinière avec une efficacité environ 40 fois plus grande que l’AAV9 après injection intraveineuse (Deverman et al., 2016).
La mise en œuvre est actuellement cédée sous licence au Cambridge startup Voyager Therapeutics in Massachusetts, qui met également au point une thérapie génétique pour la SLA SOD1. Voyager Therapeutics souhaite déposer, à la fin de 2017, une demande d’IND (investigational new drug) pour leur stratégie de traitement potentiel pour la SLA, appelée VY-SOD101.
Une question essentielle sera de savoir si le SOD1 est nécessaire pour réduire les niveaux de radicaux libres qui se forment dans les principaux tissus touchés par la maladie. Par conséquent, un nombre croissant d’équipes de recherche élaborent des stratégies du type ‘ effacer et remplacer’ qui visent à réduire le SOD1 mutant et, en même temps, produire l’enzyme naturelle pour aider à détoxifier les principaux tissus touchés par la SLA, y compris le cerveau, la moelle épinière et les muscles.
En un ou deux coups?
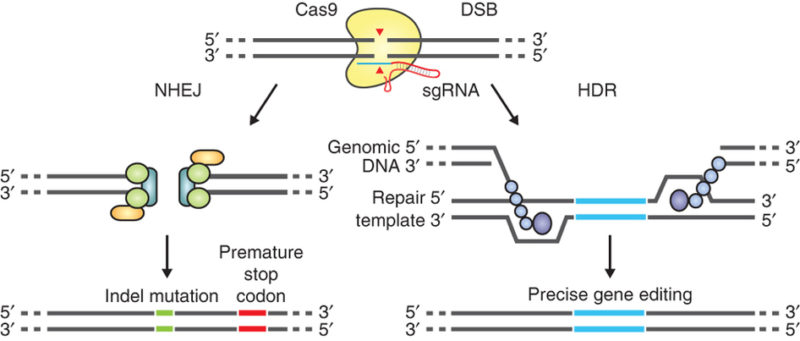
Nouvelles approches de l’ingénierie. Des chercheurs de la faculté de médecine de l’Université du Massachusetts développent une thérapie génique pour la SLA SOD1 sur base du CRISPR-Cas, qui vise à supprimer définitivement la synthèse du SOD1 mutant. Pendant ce temps, le Prof Charles Gersbach de la Duke Université élabore une stratégie de montage épigénomique, également présentée à l’AAN2017 (annual meeting of the American Academy of Neurology), qui pourrait être bénéfique à un large éventail de maladies, y compris les formes les plus courantes de la SLA, liée au gène C9orf72 (voir January 2017 conference news; Thakore et al., 2015). [Courtesy of Ran et al., 2013, Nature Publishing Group.]
Avec l’avènement des nouvelles technologies de silençage génétique, les équipes de recherche ont commencé à porter un autre regard sur leurs approches de thérapie génique et ont commencé à les modifier dans l’espoir d’optimiser leurs stratégies de lutte contre les maladies liées au SOD1. Le Dr Robert Brown s’est tourné vers la technologie d'édition de génome CRISPR/Cas9 dans l’espoir d’arrêter la synthèse du SOD1 mutant. Cette approche, dévoilée au AAN 2017, utilise des ciseaux génétiques Cas9 pour introduire des indels (insertions ou délétions) dans le gène SOD1. Le principal obstacle, selon les résultats préliminaires présentées par Zachary Taylor de l’University of Massachusetts Medical School, est d’ introduire suffisamment de Cas9 pour modifier le gène SOD1 mutant dans les motoneurones et les cellules gliales du système nerveux central. Des études précliniques sont en cours.
De l’autre coté du monde, les Dr Martine Barkats et Maria Grazia Biferi, élaborent une stratégie utilisant un nouveau silençage de gènes basé sur l’AAV10 pour s’attaquer à la SLA SOD1. Cette stratégie est maintenant optimisée et en est au stade préclinique. L’approche consiste en l’injection de la thérapie potentielle dans le sang et dans le cerveau. Avec comme objectif, selon Biferi, de s’assurer que les niveaux de l’enzyme mal repliée soient également réduits dans les tissus principaux en dehors du CNS, y compris les muscles squelettiques.
Le Dr Biferi explique : « C’est important, parce que la SLA commence à être considérée comme une maladie multisystémique. »
Actuellement, des chercheurs élaborent une stratégie similaire pour s’attaquer à la SLA C9orf72, la forme la plus courante identifiée à ce jour de la maladie.
TARgeting ALS
Un programme de détoxification pour le système nerveux central ? Les chercheurs à MeiraGTx à New York, en collaboration avec le Prof Ronald Klein et le Dr Gregory Petsko, élaborent une stratégie de thérapie génique qui vise à réduire la toxicité du TDP-43 en désintégrant l’ARNm nonsense-mediated. [Image : Emw, Wikimedia Commons.]
Pendant ce temps, le Prof Ronald Klein de la Louisiana State University aide à développer une thérapie génique qui cible plus de 95 % des cas de la maladie. L’approche, basée sur des études antérieures, dirigées par le Dr Gregory Petsko, actuellement au Weill Cornell Medical College à New York et le Dr Sami Barmada, actuellement à l’Université du Michigan, vise à réduire la toxicité de motoneurones cytoplasmique induite par TDP43 en désintégrant l’ARNm nonsense-mediated. La stratégie axée sur la thérapie génique augmente les niveaux de hUPF1, un régulateur-clé dans ce processus (voir June 2015 news; Ju et al., 2011; Barmada et al., 2015).
Cette approche, selon une étude réalisée par l’équipe du Prof Klein, prolonge la fonction motrice, au moins pendant 8 semaines dans un modèle de rats de la maladie. L’étude a révélé que le traitement potentiel, lorsqu’il est administré le jour 1, préserve la force des membres antérieurs d’un modèle de rats SLA (voir June 2015 news; Jackson et al., 2015)
Selon le Prof Klein : « C’est vraiment étonnant que l’augmentation du hUPF a cet effet protecteur vraiment particulier contre le TDP-43. Nous continuons à le constater à chaque fois. »
Comment cette stratégie parvient à atténuer la toxicité TDP-43 dans les motoneurones reste inconnu. L’approche est l’une des (au minimum) deux qui ont pour but de cibler une accumulation de TDP-43 dans le cytoplasme des motoneurones (voir April 2017 news; Becker et al., 2017).
La stratégie, dont la licence a été cédée à la startup de biotech New York MeiraGTx, en est au stade préclinique. L’évaluation de l’approche dans des modèles SLA de rats adultes se poursuit.
Traduction : Fabien
Source : The ALS Research Forum