

Ionis' derde vernieuwend antisense-medicijn voor ALS, het eerste dat ontworpen is om een brede ALS-populatie te behandelen, begint klinische studie
26-10-2020
- studie zal ION541 (BIIB105) evalueren voor de behandeling van de meeste vormen van ALS, ongeacht de familiegeschiedenis
- Tofersen en IONIS-C9Rx, gericht op erfelijke vormen van de ziekte, zijn momenteel ook in klinisch onderzoek
CARLSBAD, Californië, USA - Ionis Pharmaceuticals, de leider in antisense-therapieën, heeft vandaag aangekondigd dat de eerste patiënten gedoseerd werden met ION541 (ook bekend als BIIB105), een experimenteel antisense-medicijn dat wordt ontwikkeld als een potentiële therapie voor de behandeling van de meeste vormen van amyotrofische laterale sclerose (ALS), ongeacht de familiegeschiedenis. Dit is een nieuwe mijlpaal in de voortdurende vooruitgang van het ambitieuze programma van Ionis om nieuwe behandelingen voor ALS te ontwikkelen. Bijna alle gevallen van ALS hebben het pathologische kenmerk van TDP-43-eiwitaggregatie in motorneuronen. ION541 richt zich op het ataxine-2 RNA (ATXN2), waarvan is aangetoond dat het TDP-43-toxiciteit voorkomt of vermindert in preklinische modellen van ALS.
Vertaling: ALS Liga: Anne
Bron: PR Newswire
Van molecuul tot mens : genontdekking leidt tot experimentele therapie van de volgende generatie voor ALS
28-10-2020
Auteur : Julie Kiefer
Na meer dan 25 jaar in de maak, hebben wetenschappelijke ontdekkingen, gedaan door Stefan Pulst, MD, Dr Med, en zijn onderzoeksteam, de mijlpaal bereikt van het invoeren van klinische proeven bij mensen met een unieke behandeling voor het amyotrofisch lateraal syndroom (ALS). Het experimentele medicijn maakt deel uit van een nieuwe klasse van therapieën van de volgende generatie, die de oorzaak van de ziekte aanpakken – de genetische code – en veelbelovend zijn bij het verlichten van voorheen onbehandelbare aandoeningen.

Ontdekkingen van het laboratorium van Stefan Pulst, Dr Med, aangaande ataxie, een zeldzame ziekte, hebben geleid tot een potentiële behandeling van een andere verwoestende neurologische aandoening, ALS. Fotokrediet: Charlie Ehlert.
Het is een bemoedigende stap voorwaarts voor ALS, een snel progressief verlies van functie van cellen in het zenuwstelsel. Tegenwoordig hebben patiënten een gemiddelde levensverwachting van slechts twee tot vijf jaar na de diagnose, en geen enkele medicatie kan de ziekte vertragen of stoppen.
‘’Dit is een radicale verandering’’ zegt Pulst. ‘’Het is de eerste verbinding in zijn soort voor alle soorten ALS, genetisch en sporadisch. We hopen dat we het er over 10 jaar over zullen hebben zoals we vandaag gerichte therapieën voor kanker doen.’’
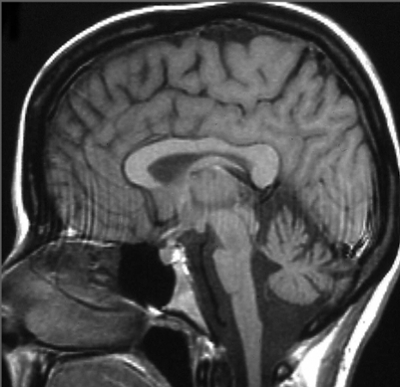
MRI-beeld met cerebellum en hersenstam atrofie bij een patiënt met Spinocerebellaire Ataxie type 2.
In de jaren negentig kon Pulst niet geweten hebben dat het ontmoeten van een gezin met een andere zenuwstelaandoening – ataxie – een pad zou openen naar het nieuwe therapeutische proefonderzoek voor ALS. Toen hij van een medische collega hoorde over het gezin, vloog Pulst van het Cedars-Sinai Medical Center in Los Angeles, waar hij voorzitter van de neurologie was, door het land naar de staat New York om familieleden te onderzoeken en monsters te verzamelen voor analyse. De ontmoeting leidde tot een vijfjarig onderzoek dat culmineerde in de ontdekking van veranderingen in een gen, ataxine-2, dat hun ‘familievloek’ veroorzaakte, nu Spinocerebellaire Ataxie type 2 genoemd. Dezelfde genetische aandoening werd ook aangetroffen in niet-verwante families rond de wereld.
Een decennium later verhuisde Pulst naar U of U Health als voorzitter van de afdeling Neurologie, waar hij onderzoek deed om een diepgaand inzicht te krijgen in wat er misgaat in het lichaam tijdens het verloop van de ziekte. Met zijn naaste medewerker, Daniel Scoles, universitair hoofddocent neurologie, ontwikkelde hij muizen met een ataxie-achtige aandoening, die een middel verschaften om genen en biochemische paden te ontdekken die na verloop van tijd verstoord raken. Het ziektemodel speelde ook een tweede kritische rol; omdat het het gemuteerde menselijke ataxine-2-gen tot expressie bracht, werd het een hulpmiddel voor het testen van gengerichte therapieën.

Onderzoek door Stefan Pulst, Dr Med en Daniel Scoles, PhD, heeft aangetoond dat geavanceerde therapie een ALS-gelijke aandoening in dieren omkeert. Fotokrediet: Charlie Ehlert.
Pulst en Scoles werkten samen met Ionis Pharmaceuticals om een nieuwe behandelingsaanpak na te streven die veelbelovend begon te worden voor andere neurodegeneratieve aandoeningen. Ze toonden aan dat een type medicijn, bekend als antisense, de expressie van het ataxaine-2-gen verminderde en de progressie van de ataxie-achtige toestand van de dieren vertraagde. Muizen presteerden beter in balans- en coördinatietesten en vertoonden tekenen van herstel op cellulair niveau. Het antisense-medicijn is een chemisch gesynthetiseerd DNA-achtig molecuul dat zich richt op ataxine-2-mRNA, wat resulteert in de vernietiging ervan.
Bij een verrassende bevinding, toonde een tweede studie aan dat een vergelijkbaar antisense-medicijn tegen ataxine-2 ook de gezondheid verbeterde van muizen met een ALS-achtige aandoening. In dit geval was het effect indirect en verminderde de toxische klontering van een ander eiwit waarvan bekend is dat het betrokken is bij het veroorzaken van ALS. De behandeling verlengde het leven van de ALS-muizen aanzienlijk.

De klinische testen zijn een mijlpaal gestoeld op decennia onderzoek. In beeld, leden van het Pulst laboratorium, Sharan Paul, PhD, en Warunsee Dansithong, PhD. Fotokrediet: Charlie Ehlert.
‘’Dit was een conceptbewijs dat deze verbindingen de basis zouden kunnen worden voor nieuwe therapieën voor neurodenegeratieve ziekten, die tot nu toe grotendeels ondoordringbaar waren’’, legt Pulst uit. Met steun van de National Institutes of Health, hebben Pulst, Scoles en Ionis Pharmaceuticals de behandeling verder verfijnd en een antisense-medicijn gevonden dat goed presteerde in dierstudies met een ongeëvenaarde veiligheid en effectiviteit. Op basis van deze resultaten, gaf de Amerikaanse FDA toestemming voor proeven bij mensen. Fase 1 klinische veiligheidsproeven van het medicijn, BIIB105 genaamd, worden gesponsord door Biogen en beginnen op 2 september met het inschrijven van patiënten.
‘’Eén van mijn professoren vertelde me dat het zeldzaam is dat iemand een betekenisvol verschil maakt in de wetenschap’’, zegt Scoles. ‘’Maar, we zijn nooit gestopt met proberen. Hopelijk maakt dit medicijn een significant verschil.’’
Vertaling: Gerda Eynatten-Bové
Bron: University of Utah Health