

Stamt ALS af en verspreidt het zich?
13-12-2012
Auteur: John M. Ravits, M.D. Professor of Clinical Neuroscience, Head ALS and Neuromuscular Translational Research University of California, San Diego
Een van de mysteries van ALS is dat de ziekte diverse gedaanten aanneemt bij pALS onderling. Voor elke pALS schijnt er een verschillend geheel van problemen en uitdagingen te bestaan. Sommigen kampen met spreekproblemen, sommigen met stapproblemen, sommigen met armproblemen, sommigen met de rest van hun lichaam. Nu wordt ook al erkend dat sommige pALS problemen hebben met taal, gedrag of zelfs met de eigen persoonlijkheid. Dat kan je gemakkelijk nagaan wanneer je contact neemt met een patiënten hulpgroep of een gespecialiseerde website.
Hoe kan dat? Hoe kan eenzelfde ziekte zo verschillen van pALS tot pALS? Wat is gemeenschappelijk en dus een onderdeel van eenzelfde diagnose die zo divers verdeeld is tussen de pALS?
De medische term voor die schijnbare verschillen luidt: heterogeniteit door fenotype. Bij ALS houdt dat in: een verschil in de locatie van de zwakte – bijvoorbeeld, spreken en slikken (bulbaire vorm), armen of benen – en in de aard van de zwakte – bovenste motorische neuronen wat stijfheid en spasticiteit veroorzaakt en lagere motorische neuronen wat verlamming en atrofie of beiden veroorzaakt.
Onderliggend aan het verschijnen van zwakte is degeneratie of verdwijning van motorische neuronen in het centraal zenuwstelsel. Dat degeneratieproces selecteert op een of andere wijze motorische neuronen boven alle andere neuronen als doel. Onderliggend is dus de degeneratie van motorische neuronen waar ook in het centraal zenuwstelsel zij zich bevinden en welke problemen ook zij veroorzaken; het fundamentele kenmerk is motorische neuronen degeneratie en de daaruit volgende verlies aan motorische functie.
Hoe verklaart dit dan heterogeniteit door fenotype? Motorische neuronen bevinden zich in specifieke anatomische locaties in de hersenen, de hersenstam of het ruggenmerg. De locatie in het zenuwstelsel bepaald welke spieren in welk deel van het lichaam gecontroleerd worden en de manier waarop die controle uitgeoefend wordt. Het motorisch systeem is dus een netwerk van elektrische circuits voor de spieren. Om die diagrammen met ‘bedrading’ te begrijpen werkten klinische neurologen, neuro-anatomici, neuropathologen en neurofysiologen in Duitsland, Oostenrijk, Rusland, Engeland, Australië, Italië, Spanje en Frankrijk meer dan 50 jaren in de tweede helft van de negentiende eeuw. En terwijl dat gebeurde werd ALS, de ziekte die dat het systeem aanviel, herkent en begrepen. Bij het ophelderen van de organisatie van het motorisch systeem en van ALS, de bedreiging ervan, werden die beide definitief met elkaar verbonden.
Recent stelden een aantal neurologen in diverse landen zich nieuwe vragen over het feit of die twee gehelen integraal met elkaar verbonden zouden zijn. Bij het uitbreken van ALS, voordat het geheel te complex wordt, kunnen klinische neurologen door eenvoudig klinisch onderzoek al uitzoeken waar in het zenuwstelsel de falende motorische neuronen gelokaliseerd zijn. Zij kunnen lokaliseren waar in het netwerk problemen resideren. Er worden nu twee uitgesproken fenomenen besproken: (1) ALS begint heel focaal (in een welbepaalde aard) totaal willekeurig in het motorisch circuit en (2) ALS lijkt zich progressief naar buiten toe te verspreiden vanuit die locatie door anatomische padtrajecten te volgen, ofwel naar een naastgelegen regio of binnen specifieke netwerken.
Merkwaardig is dat dit meebrengt dat ALS zich verspreidt! Het mag dan aanvankelijk focaal zijn, inwerken op beperkte regio’s van de hersenen of van het ruggenmerg, maar dan gaat het verder of spreidt het zich uit naar buiten toe om het anatomisch uit te drukken door het rekruteren of het inpalmen van motorische neuronen in de onmiddellijke nabijheid. Dus, de heterogeniteit door fenotype wat zo intrigeert zou kunnen uitgelegd worden door (1) de specifieke omgeving in het zenuwstelsel waar het uitbreken begint, en dat kan willekeurig waar zijn; (2) de mate waarin de bovenste motorische neuronen en de lagere motorische neuronen betrokken zijn, en dat is ook variabel; (3) het padtraject van uittredende verspreiding en dat hangt af van de exacte locatie.
Ideeën rond verspreiding waren ter discussie op het domein van neurodegeneratie tijdens de laatste vijf of tien jaren. Alzheimer’s en Parkinson’s bijvoorbeeld zijn twee degeneratieve ziekten die met ALS het feit delen dat bepaalde populaties van neuronen selectief kwetsbaar zijn. Bij Alzheimer’s en Parkinson’s werd gedacht dat het concept van verspreiding optreedt in een stereotypisch patroon van aanvang tot einde van de ziekte. In vergelijking daarmee lijkt ALS anders te zijn, omdat blijkbaar de uitbraak plaatsvindt in gelijk welke locatie ergens in het motorisch systeem. Het verschil is dan onomstotelijk aanwezig ofwel is het er omdat het aspect van verspreiding niet gemakkelijk geobserveerd kan worden in de andere twee ziekten. Zijn dus Parkinson’s en Alzheimer’s verschillend in dit opzicht? Of is het zo dat ALS ons een unieke kijk biedt op neurodegeneratie? Dat zijn de puzzelstukken die op dit moment besproken en onderzocht worden.
De onmiddellijke te beantwoorde vraag over ALS is dan: doet dat zich voor en hoe geordend is het? Of zijn er andere verklaringen, zoals genetische- en ontwikkelingsprogrammering, die beter het ontstaanspatroon en de progressie voor elke pALS zou kunnen verklaren. Er is een sleutel gevonden bij familiale ALS (fALS), waar een specifiek gendefect de ziekte veroorzaakt. Die genmutatie bevindt zich in iedere cel van het lichaam, niet alleen op een enkele locatie of in een enkel celtype. Het is duidelijk dat er waarneembare fenotype heterogeniteit is binnen de familie, waarschijnlijk vergelijkbaar met de heterogeniteit die voorkomt bij sALS. Sommige familieleden hebben bulbaire ALS, bij anderen begint ALS in de armen en bij anderen in een been en in de meeste gevallen verspreid de ziekte zich van daaruit naar buiten. Dat betekent dat het genetisch defect de uitbraak van ALS veroorzaakt op een welbepaalde anatomische locatie die dan het fenotype bepaalt.
Hoe kan verspreiding voorkomen op een cellulair en moleculair niveau? Er zijn veel verschillende mogelijkheden. Een manier is dat de cellulaire micro-omgeving van de neuronen aangetast wordt door de ziekte en lokale veranderingen een uitstroom van gebeurtenissen die naar buiten vloeien in gang zetten. Een aantal verschillende cellen zullen daar steeds bij betrokken zijn: astrocyten en micorglia cellen, zoals ook recentelijk bekend oligodendrocieten, alle cellen die vitale functies naast neuronen vertolken. Een andere manier die opgedoken is betreft toxisch oplosbare factor(en) die zich kan/kunnen verspreiden doorheen het zenuwstelsel en die een waarneembare toxische uitstoot opwekt naar buiten toe. En vele jaren reeds zijn belangrijke vouweigenschappen van eiwitten onder intensieve studie in wat prionen biologie genoemd wordt, de sleuteleigenschap daarvan is dat een eiwit verkeerd ontvouwen kan worden en dat het misvouwen eiwit andere kopieën opwekken die op dezelfde manier misvouwen zijn en die zich op die manier verspreiden in wat prion-achtige uitstroom ( prion-like propagation) genoemd wordt.
Verspreiding heeft belangrijke therapeutische implicaties! Eerstens, indien wij het proces van verspreiding zouden kunnen begrijpen, zouden wij in staat zijn om die therapeutisch aan te sturen; daarbij zou dan het proces aangevallen worden dat zelf de cel aanvalt.
Tweedens, wanneer wij het fenomeen vroeg genoeg zouden kunnen vastgrijpen, zouden wij het regionaal kunnen bestrijden. Geneesmiddelen dringen overal door om regio’s te bereiken waar zij nodig zijn. Maar regionale strategieën zijn verschillend: zij zijn lokaal. Dat verschil is vergelijkbaar met het gebruik van systemische antibiotica of van een lokale zalf om een infectie te behandelen. Nergens is dat verschil meer uitgesproken dan met het geval van de huidige stamceltherapieën. In een therapeutische strijd die gevoerd wordt door stamcellen, dienen die geplaatst te worden in strategische regio’s in het zenuwstelsel. Zij kunnen niet systemisch toegediend worden. Maar welke zijn “strategische regio’s”? Zijn dat regio’s waarin de ziekte het meest actief is? Of alternatief, zijn dat regio’s tot waar het ziektebeeld nadert maar nog niet heeft toegeslagen (in een poging om die te verdedigen en tegelijkertijd de andere regio’s te onderwerpen)? Dat moet allemaal nog vastgelegd worden terwijl die therapieën zich ontwikkelen.
Gedurende het volgende aantal jaren zal ziekteverspreiding in toenemende mate bestudeerd worden en dat zal ons ongetwijfeld leiden naar nieuwe inzichten, en dat zal ons dichter brengen bij het wereldwijde doel van een directe, fundamentele therapie om ALS te bestrijden.
Afbeelding
Een model van ALS-progressie gebaseerd op een focale start gevolgd door verspreiding
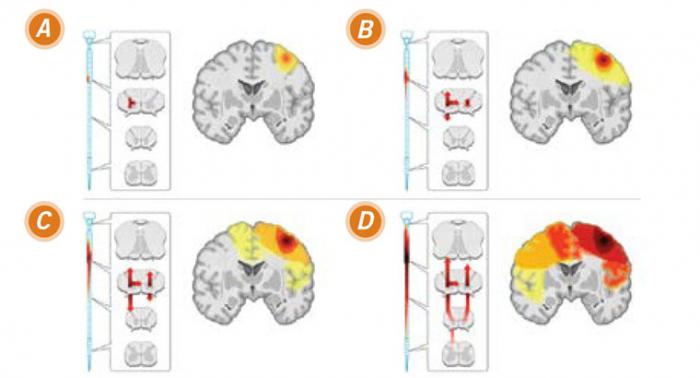
Image courtesy of Ravits, J. M. et al. Neurology 2009;73:805-811
A: Aanvang: Bij de klinische start, de degeneratie betreft motorische neuronen in de hersenen en het ruggenmerg die hetzelfde periferische lichaamsdeel (hier op de afbeelding de rechterhand) activeert.
B: De vroegste verspreiding: over een tijd zal het ziekteproces zich verspreiden doorheen de hersenen en het ruggenmerg en zullen de klinische sporen daarvan toenemen.
C: Volgehouden verspreiding naar buiten: Na verloop van tijd begint de ziekte meer complex te worden.
D: Vergevorderde verspeiding: Uiteindelijk zal het ziekteproces er diffuus en symmetrisch gaan uitzien, de details daarvan hangen af van de aanvang.
Vertaling: ALS Liga: Dirk
Bron: Research ALS Today