

Chercheurs identifient l'agent provocateur qui entraînerait la mort des neurones chez les patients atteints de SLA
30-08-2016
Harvard Medical School
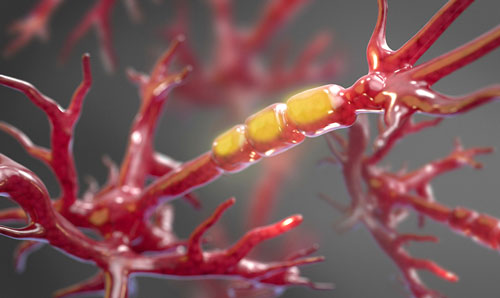
IMAGE: Une matière molle et ressemblant au gel, appelée myéline, entoure les axones des neurones afin de les protéger contre les endommagements. Dans les patients atteints de SLA la gaine protectrice est dépouillée graduellement et fait que les axones sont dénudés, exposés et vulnérables aux dégats. Les chercheurs de Harvard Medical School ont identifié un provocateur principal de ce processus.
Crédits: non applicable : image de stock
Des scientifiques de la Harvard Medical School ont identifié un agent provocateur essentiel dans les dégâts causés aux neurones chez les personnes atteintes de sclérose latérale amyotrophique ou SLA, un désordre neurodégénératif progressif et inguérissable.
Les chercheurs disent que les résultats de leur étude, publiée le 5 août dans le journal Science, pourraient mener à la découverte de nouvelles thérapies pour mettre fin à la progression de cette maladie uniformément fatale et qui affecte plus de 30.000 Américains. Actuellement un tel traitement est développé pour être testé sur les humains et ce après que la présente étude montrait qu'il pouvait arrêter la dégradation des neurones dans les souris atteintes de SLA.
Les premières manifestations de SLA sont marquées par la dégradation progressive et finalement la mort des axones de neurones, ces aspérités délicates sur les neurones qui transmettent des signaux d'une cellule à l'autre.
L'étude HMS révèle que le comportement aberrant d'un enzyme appelé RIPK1 occasionne des dégâts aux axones des neurones en dérangeant la production de la myéline, la substance molle ressemblant au gel qui enveloppe les axones dans le but de les protéger contre les blessures.
"Non seulement notre étude explique du nouveau sur les mécanismes causant les blessures et la mort des axones mais en outre elle identifie une stratégie qui pourrait, en guise de protection, réduire leur dégradation en affaiblissant l'activité de RIPK1," disait le chercheur principal de l'étude Junying Yuan, le Professeur de Biologie Cellulaire Elizabeth D. Hay de la HMS.
Les nouvelles trouvailles suivent de près une série de découvertes faites par Yuan et ses collègues durant la décennie passée qui ont marqué RIPK1 comme un agent provocateur principal de l'inflammation et de la mort des cellules. Mais jusqu'à l'heure actuelle les scientifiques ne connaissaient pas son rôle quant à la mort des axones et SLA.
Les tests conduits dans les cellules SLA des souris et des humains révèlent que, quand l'enzyme RIPK1 est hors de contrôle, elle pourrait causer des dégâts aux axones en déclenchant une réaction en chaîne chimique qui culminerait dans l'enlèvement de la myéline protectrice des axones et la mise en marche de leur dégénération --le trait principal de SLA.
Les chercheurs ont trouvé que RIPK1 cause des dégâts en attaquant directement dans le corps les endroits où la myéline est produite --des neurones connus sous le nom d'oligodendrocytes, qui sécrètent une substance molle, riche en graisse et protéine, qui va envelopper les axones pour supporter leur fonction et les protéger contre les blessures.
Partant du travail précédent du labo de Yuan qui démontrait que l'activité de RIPK1 pourrait être bloquée par un agent chimique appelé necrostatin-1, l'équipe de scientifiques a fait des tests pour savoir comment les cellules SLA cultivées au labo réagiraient au même traitement. En effet, le necrostatin-1 refoulait l'activité de RIPK1 dans les cellules de souris qui avaient été altérées génétiquement pour développer SLA.
Dans une dernière série d'expériences les chercheurs avaient recours au necrostatin-1 pour traiter les souris ayant des déficiences aux axones et des pattes de derrière faibles, un signe distinct de la détérioration des axones semblable à la faiblesse des muscles présente lorsque la SLA commence à se manifester chez les humains. Non seulement necrostatin-1 réparait l'enveloppe de myéline et arrêtait les dégâts causés aux axones mais il empêchait l'affaiblissement des membres des animaux traités.
Relier les Points
Au début de leurs tests les chercheurs s'intéressaient particulièrement au gène appelé optineurine (OPTN). Des recherches passées ont révélé des déficits d'OPTN chez les personnes atteintes de SLA, tant les formes héritées que sporadiques, mais les chercheurs n'étaient pas sûrs si et comment OPTN faisait partie du développement de la maladie. Pour atteindre plus de sûreté les chercheurs créaient des souris qui étaient tellement altérées génétiquement qu'elles n'avaient pas OPTN. En examinant les cellules de la moelle épinière sous le microscope, les chercheurs constataient que les axones des souris n'ayant pas ce gène OPTN étaient gonflés et enflammés et ils étaient moins nombreux que dans les cellules de moelle épinière trouvées dans les souris ayant le gène OPTN. En outre ces axones faisaient preuve d'une dégradation de la myéline. Chose remarquable: les chercheurs constataient la présence des mêmes signaux de la mort des axones dans les cellules de la moelle épinière obtenues de patients humains atteints de SLA. Les souris présentant ce déficit d'OPTN faisaient également preuve de perte de force dans les pattes de derrière. Les tests suivants révélaient que le manque d'OPTN était nuisible en particulier aux cellules sécrétant la myéline. Par conséquent les chercheurs concluaient que le déficit d'OPTN paralysait directement la fabrication de myéline dans le système nerveux. Toutefois il restait la question de savoir comment l'absence d'OPTN cause des dégâts à ces cellules?
Un Fusil Fumant
Dans leurs recherches de matières chimiques généralement présentes durant l'inflammation et la mort des cellules, les scientifiques ont constaté des niveaux anormalement élevés de RIPK1--un agent promoteur connu de la mort des cellules--dans les cellules de la moelle épinière de souris qui n'ont pas OPTN. En outre les chercheurs observaient des traces d'autres agents chimiques destructeurs souvent recrutés par RIPK1 pour tuer des cellules.
"C'était comme si nous voyions les empreintes chimiques de la mort de cellules délaissées par RIPK1 et ses recrutés," disait Yuan.
Et Yuan ajoutait que cette observation était le fusil fumant qui liait l'inconduite de RIPK1 au déficit d'OPTN. En d'autres termes et toujours selon les chercheurs: quand le gène OPTN fonctionne comme il faut, il semble régler le comportement de RIPK1 en assurant que ses niveaux sont contrôlés de près, qu'il se dégrade vite et qu'il est chassé des cellules à temps. Toujours est-il qu'en l'absence d'une telle omission de RIPK1 on aura l'impression qu'il perd tout contrôle et donc qu'il va causer des dégâts.
Dans une série limitée de tests les checheurs examinaient des neurones qu'ils avaient obtenus de souris qui étaient atteintes de la forme héréditaire de SLA la plus courante, causée par des mutations dans un gène appelé SOD1. En effet, les niveaux de RIPK1 étaient élevés dans ces cellules aussi. Par conséquent les chercheurs ont constaté que peut-être OPTN n'est pas le seul gène qui règle le comportement de RIPK1. Au contraire, RIPK1 semble stimuler les dégâts causés aux axones et ce dans différentes formes de SLA, tant héritées qu'acquises.
Les trouvailles suggèrent que RIPK1 pourra jouer un rôle dans toute une rangée de maladies neurodégénératives marquées par des dégâts causés aux axones, y inclus la sclérose en plaques, certaines formes d'atrophie spinale musculaire et même la maladie d'Alzheimer.
Le Harvard Office of Technology Development (OTD) et certaines institutions qui collaborent avec ont développé un ensemble de brevets pour des produits modulaires de RIPK1. Harvard OTD a donné une licence à une compagnie biotechnologique.
Traduction : André De Laet
Source : EurekAlert
![]()
RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS