

Mise à jour du projet se rapportant à la SLA Interactome: différentes protéines de la SLA et des mécanismes pathologiques aboutissant à la SLA
23-01-2017
Alors que nous savons de plus en plus en matière de génétique de la SLA, nous connaissons finalement très peu sur les protéines liées à celle-ci. Due à une déformation du gène de la SLA, la cellule nerveuse comporte trop peu ou trop de protéine déformée. Ce projet ‘interactome SLA’ nous fournit un terrain d’étude sur les protéines se liant à celles de la SLA. Toutes les protéines unies à d’autres bien spécifiques forment le “interactome”. De plus amples connaissances sur les protéines de la SLA et la manière dont elles se lient peuvent fournir des indications importantes en matière de possibilités futures de traitement de la SLA.
Les résultats de ce projet démontrent qu’il existe différentes formes de SLA. Au niveau de celle-ci, des problèmes avec ces protéines dans les cellules nerveuses motrices surviennent, mais au sein de différents groupes de patients atteints par la maladie, il semble que les différentes espèces de protéines de la SLA sont à l’origine de ces problèmes.
Accumulation de protéines au niveau du neurone.
En cas de patients touchés par la SLA, des agrégats, agglutinations ou accumulations de protéines amassées dans le neurone se distinguent. Les protéines doivent en principe remplir leur rôle auprès de la cellule mais au lieu de cela, elles sont collées dans l’agrégat. Les analyses effectuées à l’occasion de cette étude ont révélé plusieurs protéines inconnues par le processus pathologique de la SLA.

Prof. Jeroen Pasterkam : “Nous avons examiné à quelles autres les 6 protéines de la SLA s’étaient liées afin d’obtenir une vue plus perspicace au sujet des mécanismes de la maladie. Nous avons découvert qu’il existait un grand chevauchement des protéines qui se lient au FUS, TDP-43 et ATXN2 et celles concernées par l’ OPTN et l’ UBQLN2”.
Analyse des protéines qui se lient entre elles
Pasterkamp éclaircit: Nous avons découvert que pour 3 protéines différentes chez les patients souffrant de SLA (FUS, TDP-43 en ATXN2) celles-ci se lient aux mêmes. Cela implique que ces trois protéines provoquent de la même manière la SLA. Les protéines qui se lient aux FUS, TDP-43 et l’ATXN2 paraissent vraisemblablement avoir leur importance lorsqu’il s’agit de traiter l’ARN.
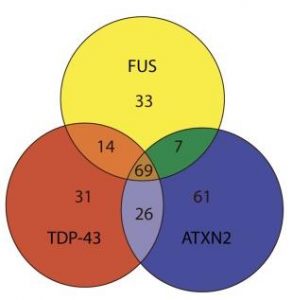 Figure 1 : Ce schéma révèle 69 sortes de protéines se liant aussi bien aux FUS-, TDP-43 qu’aux protéines ATXN2. Cela veut dire que chez les patients atteints par la SLA comprenant des mutations dans les FUS, TDP-43 ainsi que dans les gènes ATXN2, un processus comparable de la maladie a lieu dans les neurones moteurs, dans lesquels la même sorte de protéines se fixe à celles concernées par la SLA. A cause de ces agrégats de protéines, le neurone est déréglé et finit par succomber
Figure 1 : Ce schéma révèle 69 sortes de protéines se liant aussi bien aux FUS-, TDP-43 qu’aux protéines ATXN2. Cela veut dire que chez les patients atteints par la SLA comprenant des mutations dans les FUS, TDP-43 ainsi que dans les gènes ATXN2, un processus comparable de la maladie a lieu dans les neurones moteurs, dans lesquels la même sorte de protéines se fixe à celles concernées par la SLA. A cause de ces agrégats de protéines, le neurone est déréglé et finit par succomber
Les protéines de la SLA, l’OPTN et le UBQLN2 sont tout aussi bien examinées. Celles-ci entrainent également des caillots dans le neurone mais il s’agit d’autres protéines qui s’y lient que celles mentionnées auparavant : FUS, TDP-43 et ATXN2. Des déformations dans OPT et UBQLN2 peuvent également être la cause de la SLA, mais cela se passe par un autre processus de la maladie. Nous sommes d’avis que les protéines dans la figure 1 et celles dans la figure 2 connaissent chacune leur ‘disease pathway’ (leur trajectoire vers la SLA). Par conséquent, plusieurs processus de la maladie mènent à la SLA.
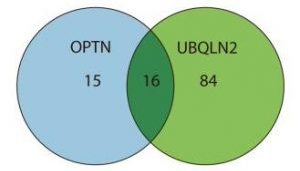 Schéma 2 : Dans le schéma ci-dessus, veuillez noter que 16 formes de protéines se lient aussi bien à l’ OPTN qu’ à la UBZLN2. Cela signifie que chez les patients atteints par la SLA comportant des mutations dans les gènes OPTN et UBQLN2 un processus comparable de la maladie sconcerne e des cellules nerveuses motrices, où la même sorte de protéine se colle à celles de la SLA.
Schéma 2 : Dans le schéma ci-dessus, veuillez noter que 16 formes de protéines se lient aussi bien à l’ OPTN qu’ à la UBZLN2. Cela signifie que chez les patients atteints par la SLA comportant des mutations dans les gènes OPTN et UBQLN2 un processus comparable de la maladie sconcerne e des cellules nerveuses motrices, où la même sorte de protéine se colle à celles de la SLA.
Les itinéraires de la maladie
Les symptômes chez le patient se présentent de la même façon, majoritairement par l’accentuation de la déficience par l’extinction des cellules nerveuses motrices. Mais la raison qui fait que les cellules nerveuses meurent peut varier d’un patient à l’autre. Pasterkamp: “A partir de cette recherche, nous croyons que chez certains patients les protéines FUS, TDP-43 et ATXN sont responsables, tandis que chez d’autres les protéines OPTN et UBQLN2 sont la cause”.
Ces résultats prouvent que l’‘interactomics’, l’analyse des protéines qui se lient les unes ou autres durant l’évolution de la maladie dans la cellule nerveuse, encourage la compréhension au niveau du processus de la maladie de la SLA et pour distinguer les différents ‘itinéraires engagés par la maladie’.
Une médecine personnalisée
Le nouveau projet TryMe utilise ces résultats par le biais des lignées de cellules souches pour démontrer comment les neurones protéines SLA ne fonctionnent pas convenablement au niveau des neurones. On recherche des traitements pour plusieurs mécanismes liés à la maladie. Au lieu d’un médicament global qui fonctionne pour tous les patients souffrant de la SLA, diverses possibilités de médecines personnalisées existent. Nous sommes à la recherche de plusieurs traitements destinés à différents groupes de patients atteints de SLA afin d’en finir avec la maladie.
Traduction : Eric Kisbulck
Source : ALS Centrum Nederland