

Des scientifiques créent des ‘graines’ protéiques qui déclenchent les principales caractéristiques pathologiques de la SLA et de la démence frontotemporale.
02-04-2025
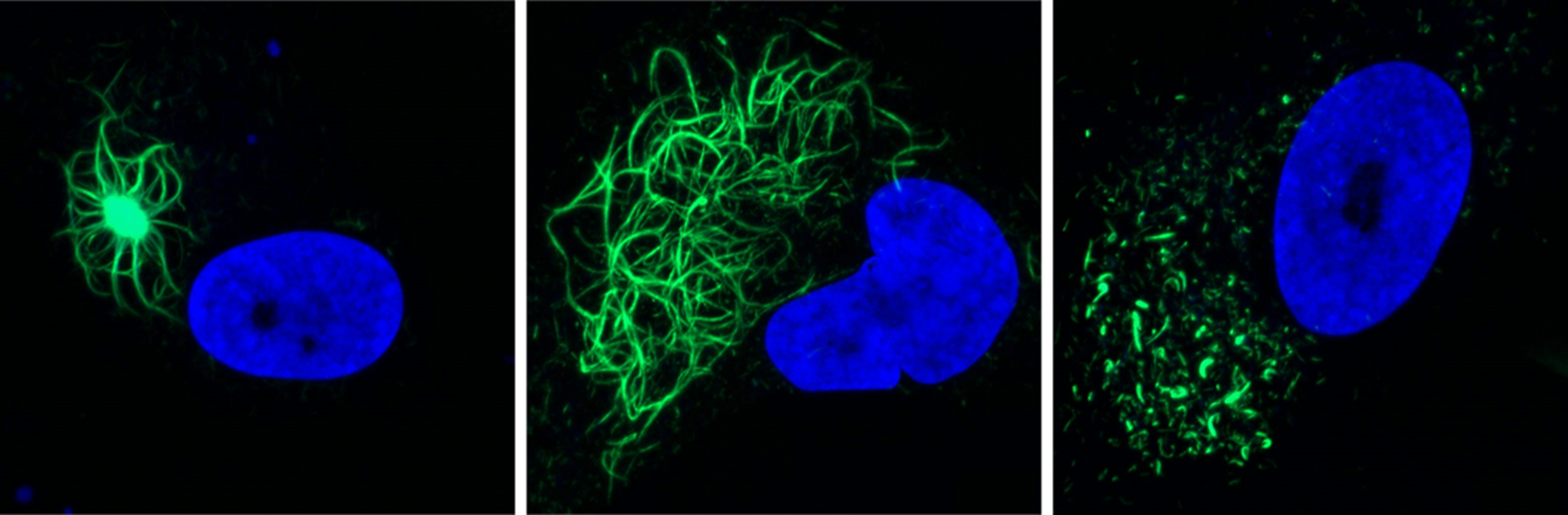
Trois types d'agrégats (colorés en vert) : des agrégats compactés contenant un noyau dense au centre (à gauche), des agrégats se présentant sous la forme d'un réseau de filaments (au centre) et des agrégats fragmentaires. Le noyau est coloré en bleu.
L'accumulation d'une protéine appelée TDP-43 est une caractéristique clé de la SLA et de la démence frontotemporale. Dans une étude récemment publiée, des chercheurs rapportent que cette accumulation a été « ensemencée » par des fragments de la protéine responsable créés en laboratoire. Ces résultats apportent des preuves supplémentaires d'un modèle de type prion, où l'agrégation protéique se produit selon un modèle. Cette avancée offre à la recherche un puissant outil pour modéliser et étudier les mécanismes à l’origine de la neurodégénérescence.
La protéine de liaison à l'ADN TAR 43, plus connue sous le nom de TDP-43, est une protéine présente dans presque toutes les cellules humaines, où elle joue un rôle essentiel dans la régulation de l'expression des gènes, le traitement de l'ARN et les réponses cellulaires au stress. Dans des conditions normales, TDP-43 contribue au maintien de la santé et du fonctionnement des neurones en contrôlant l'activation ou la désactivation des gènes et la traduction de leurs messages en protéines.
Cependant, TDP-43 est tristement célèbre pour son rôle dans plusieurs maladies neurodégénératives, notamment la SLA et la démence frontotemporale, mais aussi la maladie d'Alzheimer.
Du noyau au cytoplasme
‘’La pathologie TDP-43 est considérée comme une caractéristique déterminante de la quasi-totalité des cas de SLA et d'environ la moitié des cas de démence frontotemporale’’, explique la professeure Sandrine Da Cruz, cheffe de groupe au Centre de recherche sur le cerveau et les maladies de la VIB-KU Leuven. ‘’Dans le cerveau de ces patients, TDP-43 se localise mal, s'accumule dans le cytoplasme où il forme des inclusions insolubles et est éliminé du noyau.’’
Malgré son rôle crucial, les processus exacts à l'origine du dysfonctionnement de TDP-43 restent mal compris – une lacune urgente que des chercheurs comme Da Cruz s'efforcent de combler. Les ravages et la mort neuronale généralisée qui surviennent suite à une mauvaise localisation de TDP-43 sont très probablement dus à une combinaison de perturbations des activités normales de TDP-43 dans le noyau et de l'effet toxique des inclusions cytoplasmiques.
‘’Si les mécanismes sous-jacents sont encore mal compris, c'est en partie parce que nous manquons de systèmes modèles reproduisant de manière fiable à la fois la déplétion nucléaire de TDP-43 et son agrégation cytoplasmique’’, explique Da Cruz.
Semences d'agrégation
S'appuyant sur des rapports récents indiquant que du matériel cérébral autopsié de patients présentant des inclusions de TDP-43 peut induire une accumulation de TDP-43 insoluble dans des cellules et des modèles murins transgéniques, Da Cruz et son équipe ont cherché un moyen de reproduire ce que l'on appelle ‘ensemencement’ d'agrégation en laboratoire.
Dans une étude récemment publiée dans Neuron, l'équipe de Da Cruz décrit comment elle a produit des fibrilles de type amyloïde à partir d'un fragment de TDP-43 et explique que ces fibrilles déclenchent la pathologie TDP-43 dans les cellules humaines, notamment les neurones dérivés de cellules iPSC.
Les inclusions induites par les fibrilles reprennent bon nombre des caractéristiques clés observées chez les patients, explique Jens Rummens, doctorant : ‘’Les agrégats de TDP-43 induits par les fibrilles présentaient bon nombre des modifications observées également dans le cerveau des patients, notamment la phosphorylation et l’ubiquitination. De manière frappante, les agrégats étaient capables de recruter du TDP-43 endogène du noyau vers le cytoplasme’’.
Les similitudes se sont étendues à d’autres effets en aval, l’équipe ayant identifié des profils d’activité de gènes signatures auparavant liés à l’agrégation et à la perte nucléaire de TDP-43. Les agrégats eux-mêmes présentaient la même hétérogénéité morphologique que celle généralement observée chez les patients au fil du temps.
Outil de recherche
Les nouveaux résultats suggèrent fortement que la pathologie des protéinopathies à TDP-43 se propage de manière auto-modélisée et à la manière d’un prion, mais de nombreuses questions restent sans réponse. Comment le TDP-43 est-il « piégé » dans les agrégats ? De quoi sont-ils composés et comment déclenchent-ils une toxicité ? Quels « coups » supplémentaires sont nécessaires ? Quels sont les effets des mutations TDP-43 ? De l'âge ?
Plus important encore, cette nouvelle étude fournit aux scientifiques les outils nécessaires pour étudier les différents déclencheurs et leurs interactions complexes dans un système contrôlé.
Da Cruz: ‘’Nous avons développé un modèle précieux qui illustre les deux aspects de la pathologie TDP-43 : l’agrégation cytoplasmique et la déplétion nucléaire. Ce sera un atout précieux pour aider les chercheurs du monde entier à mieux comprendre les mécanismes pathologiques induits par TDP-43 et à sélectionner des candidats médicaments potentiels capables de modifier la progression de la maladie.’’
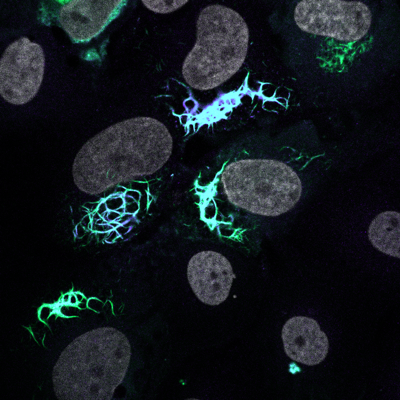
TDP-43 (vert/bleu) et noyaux (gris)
L'ensemencement de TDP-43 induit une hétérogénéité d'agrégation cytoplasmique et une perte de fonction nucléaire de TDP-43
Rummens et al. Neuron 2025
Cette recherche est le fruit d'une collaboration entre différents laboratoires du VIB, de la KU Leuven et de l'UHasselt, et a bénéficié du soutien financier de la FWO, de l'Association pour la dystrophie musculaire et de la Fondation pour la recherche sur la maladie d'Alzheimer.
Traduction: Gerda Eynatten-Bové
Source: Site web du VIB