

''ALS-omkeringen'': demografie, ziektekenmerken, behandelingen en co-morbiditeit
10-04-2018
Samenvatting
Doel: Het identificeren van verschillen in demografie, ziektekenmerken, behandelingen en co-morbiditeit (het gelijktijdig voorkomen van twee of meer aandoeningen of stoornissen bij één persoon) tussen patiënten met “amyotrofische laterale sclerose (ALS) –omkeringen” versus patiënten die lijden aan een typisch progressieve vorm van ALS.
Methoden: Mogelijke gevallen van ALS-omkeringen werden gevonden in eerdere publicaties, in de Duke ALS-kliniek, na zelfverwijzing of verwijzing door andere neurologen en in berichten op het internet. Van de 89 geïdentificeerde mogelijke omkeringen werden 36 gevallen in deze studie opgenomen gezien nazicht van het medisch dossier of literatuur hun diagnose bevestigde alsook een robuuste aanhoudende verbetering in ten minste één objectieve maatstaf (parameter). Als controlepersonen werden deelnemers aan de Pooled Resource Open-Access ALS Clinical Trials -database en het nationale ALS-register genomen. “Gevallen” en “controles” werden vergeleken aan de hand van beschrijvende statistiek.
Resultaten: gevallen van ALS-omkering waren eerder mannelijk, vertoonden een aanvang van de ziekte ter hoogte van de ledematen en evolueerden aanvankelijk sneller. De prevalentie van myasthenia gravis (MG - een aandoening gekenmerkt door spierzwakte) en zuiver lager motorneuronziekte bij “gevallen” waren hoger dan geschatte prevalenties in de algemene populatie. De kans op inname van kurkuma, luteoline, cannabidiol, azathioprine, koper, glutathion, vitamine D en visolie was groter voor “gevallen” dan voor “controles”.
Conclusies: in vergelijking met patiënten met een typisch progressieve ALS, verschilden patiënten met omkeringen qua demografie, ziektekenmerken en behandelingen. Hoewel sommigen van deze patiënten mogelijks leden aan een zeldzame antilichaam-gemedieerde, op ALS gelijkende ziekte, zoals atypische myasthenia gravis, lijkt dit uit details van hun medische onderzoeken, EMG's en familiegeschiedenis eerder onwaarschijnlijk. Onze data-analyse suggereert integendeel dat ALS-omkeringen verder onderzoek naar mechanismen van ziekteresistentie rechtvaardigen en dat meerdere behandelingen geassocieerd met ALS-omkeringen een verdere evaluatie verdienen.
Inleiding
Amyotrofische laterale sclerose (ALS) is een desastreuse en bijna universeel fatale neurodegeneratieve ziekte. Zeer zelden stopt het ziekteverloop bij een ALS patiënt en herstelt een belangrijke motorische functie. De studie van deze “ALS-omkeringen” zou een, tot nog toe te weinig onderkend, nabootsend syndroom, een genetisch mechanisme van ALS-resistentie, of een mogelijks effectieve behandeling kunnen aan het licht brengen. Wij bundelen geverifieerde gevallen van ALS-omkeringen in een databestand teneinde hun demografie, ziektekenmerken, behandelingen en co-morbiditeit te vergelijken met die van patiënten met een meer typisch progressieve vorm van ALS.
Methoden
Gegevensbronnen en onderzoekontwerp
Het betreft hier een patiënt-controle-onderzoek (vergelijkend cohort onderzoek). Potentiële gevallen werden geïdentificeerd in eerdere peer-reviewed (intercollegiaal getoetste) publicaties (n = 23), in de Duke ALS-kliniek (n = 3), na zelfverwijzing of verwijzing van andere neurologen (n = 40) en van nonpeer-reviewed berichten gepost op het internet (n = 23). Patiënten bij wie we onafhankelijk een ALS of progressieve spieratrofie (progressive muscular atrophy - PMA) -diagnose en een robuuste, stelselmatige verbetering in ten minste één objectieve maatstaf konden bevestigen aan de hand van nazicht van medisch dossier of literatuur (gedefinieerd als “ALS-omkeringen”) werden in deze studie opgenomen. Gevallen met ALS-diagnoses voldeden aan El Escorial-Revised en / of Awaji-criteria. Omkeringen werden meestal aangetoond op basis van een resolutie van denervatie zoals waargenomen op een elektromyografie (EMG), een verbeterde kracht bij handmatige spiertesten en / of een winst van ten minste 4 punten op de herziene versie van de functionele ALS beoordelingsschaal (ALSFRS-R). In sommige gevallen werden patiënten met enkel maar buitengewone verbeteringen in dagdagelijkse activiteiten opgenomen. We hebben bijvoorbeeld een geval opgenomen dat op de nadir (op het dieptepunt) niet in staat was om rechtop te staan en na haar verbetering enkele kilometers kon lopen, maar waarvoor geen formeel gedocumenteerd krachtonderzoek beschikbaar was. Patiënten met een terugval naar of onder hun eerdere dieptepunt werden uitgesloten.
Gegevens over de demografie, diagnoses, omkeringen, behandelingen en co-morbiditeit van “gevallen” werden verzameld in een databestand. “Controles” (n = 10.723) waren patiënten in de “Pooled Resource Open-Access ALS Clinical Trials (PRO-ACT) database”, gedownload op 17 oktober 2016, en vrijwillig aangeboden door PRO-ACT Consortium-leden (Prize4Life, Northeast ALS Consortium en ALS Therapy Alliance). Gegevens over demografie en familiegeschiedenis van het online, zelf-registratie portaal van het nationale ALS-register (n = 6.352) waren eveneens beschikbaar ter vergelijking.
Statistische analyse
Statistische analyses werden uitgevoerd met behulp van JMP® Pro 13.0.0. Chi-kwadraat analyses en t-tests met gepaarde steekproeven werden gebruikt om demografische en ziektekenmerken van “gevallen” te vergelijken met “controles”. Loopscoreprogressie werd verder geëvalueerd aan de hand van covariantie-analyse. Wilcoxon's rank sum testen werden gebruikt om leeftijdsgroepen te vergelijken. Behandelingen werden vergeleken met behulp van logistische regressie en Bonferroni correctie.
Goedkeuring van standaardprotocollen, registratie en toestemming van patiënten
Deze studie werd goedgekeurd door de commissie voor ethiek van Duke University. Er waren geen interventies en er werd geen beschermde gezondheidsinformatie opgenomen in de studie. Vanwege het zeer lage risico werd een verklaring van ontheffing van de geïnformeerde toestemming toegekend.
Resultaten
Diagnoses en omkeringen
In totaal werden er 36 gevallen met klinisch duidelijke ALS (n = 4), klinisch waarschijnlijke of klinisch waarschijnlijke laboratorium ondersteunde ALS (n = 23), klinisch mogelijke ALS (n = 2) en progressieve spieratrofie (PMA, n = 7) geïdentificeerd na nazicht van de literatuur (n = 16) en medische dossiers (n = 20). Informatie over hun diagnoses en omkeringen werd opgenomen in de aanvullende tabellen 1 en 2. Allen beschikten over een medische geschiedenis, neurologische onderzoeken, EMG's en opvolging voor op ALS gelijkende ziektes wat ALS diagnose waarschijnlijker maakte dan niet. Ter informatie: 19% van de gevallen had een zuiver lager motorneuronsyndroom in vergelijking met een populatieschatting van 5% van alle patiënten met motorneuronziektes. We beschouwden deze “progressieve spieratrofie” (PMA) patiënten als ALS patiënten om de eerder genoemde redenen.
Na een initiële verslechtering, was de mediane tijd tot maximale verbetering 12 maand (statistisch bereik 1- 206 maand). Er waren 16 gevallen die verbeterden bij hun laatst bekende opvolgtoetsing (follow-up). Van 18 gevallen die na hun maximale verbetering afvlakten (plateau), was de mediane duur van de opvolging 38,5 maand (statistisch bereik 3-295 maanden). De duur van de opvolging kon voor twee gevallen niet worden bepaald (deelnemers 24 en 26) omdat hun data voor maximale verbetering niet beschikbaar waren. Maatstaven toegepast om ziekteprogressie te evalueren, varieerden van geval tot geval en dit op basis van de beschikbaarheid ervan in het medisch dossier of het originele rapport. Potentiële gevallen met verbeteringen op één of meerdere objectieve maatstaven werden uitgesloten als ze vervolgens verslechterden op enige andere maatstaf. Er waren 12 gevallen met verbeteringen op de ALSFRS-R schaal (gemiddeld 9,6 punten, standaard deviatie SD 4,7 punten). De progressie van ALSFRS-R-score van het begin van de ziekte tot de laatste bekende follow-up worden weergegeven in Figuur 1. In gevallen met verbeteringen in kracht bij manuele spiertesten trad deze verbetering in de meeste gevallen op in drie of vier ledematen. Er waren negen gevallen die opnieuw een normale kracht ontwikkelden en zeven gevallen met volledige resolutie van actieve denervatie op EMG op het moment van hun omkeringen.
Demografische en ziektekenmerken
Vergelijkingen tussen de demografische gegevens en ziektekenmerken van “gevallen” en “controles” worden weergegeven in Tabel 1. Ziekteprogressie werd gemeten aan de hand van het verlies aan punten per jaar op de ALSFRS-R-loopscore, aangezien slechts weinig gevallen over volledig gedocumenteerde ALSFRS-R-scores beschikten op het moment van de nadir. Na vergelijking voor leeftijd en plaats van aantasting, bleef de loopscore progressie significant sneller in vergelijking met deze van de controles.
Co-morbiditeit
Noch PRO-ACT noch het nationale ALS-register bevatten uitgebreide informatie over de co-morbiditeit van hun deelnemers. De prevalentie van een eerdere myasthenia gravis (MG) -diagnose was echter hoger bij de “gevallen” (6%, n = 2, deelnemers 34 en 35) dan schattingen van de prevalentie bij de algemene bevolking (0,03%), hoewel ALS en MG mogelijks frequenter samen voorkomen dan kan worden voorspeld op basis van een louter toeval.
Behandelingen
Aanvullende tabel 3 bevat informatie over behandelingen die door twee of meer gevallen werden aangewend op het moment van maximale verbetering, waarbij de waarschijnlijkheid op behandeling significant groter was voor “gevallen” dan voor PRO-ACT-“controles”. Deze behandelingen, met uitzondering van azathioprine, werden uitsluitend gebruikt door gevallen die konden werden geïdentificeerd na verificatie van het medisch dossier. Na vergelijking van leeftijd en plaats van aanvang, bleken de kansen op kurkuma, azathioprine, koper, glutathion, vitamine D en visolie gebruik aanzienlijk hoger bij “gevallen” dan bij “controles”. Een gebrek aan gegevens verhinderde de vergelijking van patiënten die luteoline of cannabidiol gebruikten, maar de waarschijnlijkheid van aanwenden van deze twee behandelingen bleef groter voor “gevallen” dan “controles”, ook na controle van leeftijd en plaats van aanvang.
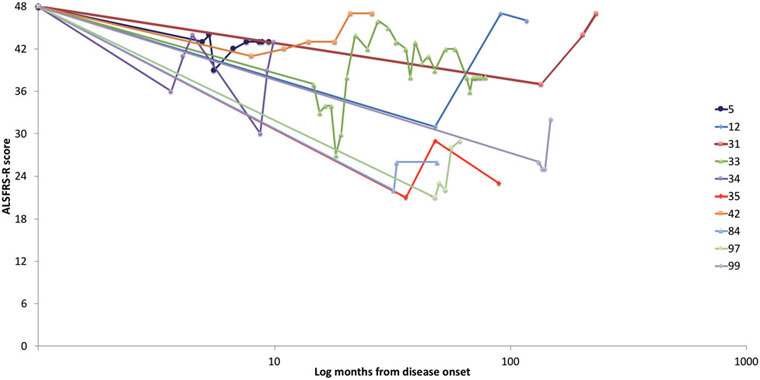
Figuur 1. ALSFRS-R-progressie. Er waren 12 gevallen met verbeteringen gemeten aan de hand van ALSFRS-R. Hiervan werden er 10 in de figuur opgenomen (geval 38 had initiële metingen aan de hand van ALSFRS voorafgaand aan de introductie van de ALSFRS-R en geval 24 had een onbekende datum van maximale verbetering). Deze grafiek toont de progressie in de loop van de tijd van deze 10 gevallen zoals gemeten door de ALSFRS-R vanaf het begin van de ziekte tot de laatst bekende follow-up. De deelnemers 5, 33, 34, 35, 42 en 99 hadden gedocumenteerde verbeteringen in aanvullende objectieve maatstaven.
ALSFRS (-R): amyotrofische laterale sclerose functionele beoordelingsschaal (-herziene versie).
Tabel 1. Demografie en ziektekarakteristieken van ALS omkeringen.
| A | Gevallen | PRO-ACT | Statistische test | Resultaat |
| Leeftijd bij aanvang | 50.1 (15.3) | 53.8 (11.8) | t = 1.89 | p = 0.0588 |
| % Mannelijk | 80.6 | 60.3 | χ2 = 6.14 | p = 0.0132 |
| % Blank | 87.5 | 95.4 | χ2 = 3.36 | p = 0.0669 |
| % Met familiale ALS historiek | 8.00 | 15.7 | χ2 = 1.10 | p = 0.2940 |
| % Aanvang in ledematen | 93.9 | 72.1 | χ2 = 7.80 | p = 0.0052 |
| Loopscore progressie | −1.59 (1.81) | −0.991 (0.762) | t = 4.28 | p < 0.0001 |
| B | Gevallen | National ALS Registry | Statistische test | Resultaat |
| Leeftijd bij diagnose | 50–59 | 50–59 | Z = 2.88 | p = 0.0040 |
| % Mannelijk | 80.6 | 60.5 | χ2 = 6.05 | p = 0.0139 |
| % Blank | 87.5 | 95.2 | χ2 = 3.08 | p = 0.0792 |
| % Met familiale ALS historiek | 4.00 | 4.20 | χ2 = 0.003 | p = 0.9594 |
ALS: amyotrophic lateral sclerosis; ALSFRS-R: ALS Functional Rating Score–Revised; PRO-ACT: Pooled Resource Open-Access ALS Clinical Trials.
In deze tabel worden beschikbare demografische gegevens en ziektekenmerken van patiënten met ALS-omkeringen vergeleken met patiënten in de PRO-ACT-database (A) en het nationale ALS-register (B). Gegevens over familiegeschiedenis in het register omvatten familieleden in de eerste graad en gegevens over de familiegeschiedenis inde PRO-ACT omvatten eerstegraads tot derdegraads verwanten. Als zodanig werden gevallen met eerste tot en met derdegraads verwanten met ALS geacht een familiegeschiedenis te hebben in A en alleen gevallen met eerstegraads familieleden met ALS werden geacht een familiegeschiedenis te hebben in B. Alleen leeftijdsgroepen waren beschikbaar in het register, en werden vergeleken met behulp van “Wilcoxon's rank sum-tests”. Mediane leeftijdsklassen waren equivalent, maar de “rank sum” van “case”-leeftijd-klassen waren significant lager en de “rank sum” van “control”-leeftijd-klassen waren significant hoger dan hun respectievelijk verwachte waarden. Verhoudingen werden geanalyseerd met Pearson's chikwadraattests en gemiddelden werden geanalyseerd met t-tests met gepaarde steekproeven. Standaardafwijkingen werden, indien van toepassing, weergegeven tussen haakjes. De progressieratio van de loopscore wordt gemeten als verandering per jaar in punten op de ALSFRS-R “loop” vraag.
Discussie
Voor het eerst werden geverifieerde gevallen van ALS-omkeringen gecompileerd in een databank ter vergelijking met patiënten met een meer typisch vorm van progressieve ALS. Er werden enkele verschillen aangetoond in demografische kenmerken en ziektekenmerken van “gevallen” in vergelijking met “controles”. Deze verschillen zijn consistent ten opzichte van twee groepen controles en komen overeen met eerder gepubliceerde gegevens over ALS-omkeringen. Het is belangrijk op te merken dat de definitie van een ALS-omkering gebruikt in deze studie verschilt van vorige definities; er werd uitgegaan van een verbetering van vier of meer punten op de ALSFRS-R schaal en dit over een tijdsspanne van ten minste 12 maand. Deze nieuwe definitie maakt de toevoeging mogelijk van patiënten met aanzienlijke verbeteringen die geen gedocumenteerde ALSFRS-R scores hebben. Eén van deze gevallen zonder ALSFRS-R score had bijvoorbeeld niet langer behoefte aan een beademingsapparaat na 17 jaar afhankelijkheid en een andere begon te lopen na een jaar van quadriplegie. Onze nieuwe definitie vereist ook dat verbeteringen robuust en duurzaam zijn, aangezien kleine, tijdelijke verbeteringen in ALSFRS-R en spierkracht niet ongebruikelijk zijn bij patiënten met een typisch progressieve ALS. Onze bekommernis met betrekking tot eerder werk met betrekking tot ALS-omkeringen aan de hand van de PRO-ACT-databank betrof moeilijkheden bij het interpreteren van verbeteringen op de ALSFRS-R schaal zonder bijhorende gegevens van een andere resulterende maatstaf en een gebrek aan informatie met betrekking tot ziekten met een ALS-nabootsing syndroom. De meerderheid van de patiënten die hier werd beschreven met verbeteringen in ALSFRS-R beschikte ook over gedocumenteerde verbeteringen gebaseerd op krachtonderzoek of andere resulterende maatstaven. Bovendien beschikten alle gevallen over informatie met betrekking tot nabootsende ziekten. Hoewel de meeste patiënten beschikten over uitgebreide beeldvorming, EMG / zenuw geleidingstudies en laboratoriumonderzoek, varieerde de beschikbaarheid ervan van geval tot geval.
Een mogelijke hypothese is dat voor een aantal van de “gevallen” een verkeerde initiële diagnose werd gesteld en er mogelijks zeer zeldzame antilichaam-gemedieerde ALS-nabootsingssyndromen in het spel waren. De verhoogde aantallen van co-morbide MG en verhoogde waarschijnlijkheid van azathioprine gebruik bij de “gevallen” kan dit mogelijks ondersteunen. Deelnemer 34, bijvoorbeeld, werd gediagnosticeerd met seropositieve MG met reactie op immuun modulatie. Bij hem werd ALS twee jaar later gediagnosticeerd na het ontwikkelen van toenemende voetzool reflexen, pseudo bulbair effect en stevige peesreflexen in de armen en benen met tekenen van denervatie en reïnnervatie op EMG bij afwezigheid van herhaalde stimulatie van de zenuwen. Deelnemer 35 werd gediagnosticeerd met MG na een subacute episode van ptosis, dysartrie, dysfagie en zwakte die binnen de 2 maand volledig was verdwenen. Hij werd dertig tot veertig jaar later gediagnosticeerd met PMA op basis van wijdverspreide atrofie, zwakte (meer distaal in de bovenste ledematen) en tekenen van denervatie en reïnnervatie op EMG. MG en ALS zou echter vaker samen kunnen voorkomen dan enkel op basis van louter toeval zou worden verwacht. Het ontbreken van fluctuerende of oculaire zwakte, de aanwezigheid van bovenste motorneuron signalen en de aanwezigheid van EMG-denervatie in de meeste van onze gevallen, leveren hiervoor sterke argumenten die louter op basis van MG alleen niet verklaard kunnen worden. Het vergelijkbare percentage ALS-familiegeschiedenis in onze “gevallen” en “controles” is verder in tegenspraak met de mogelijkheid dat sommige van deze gevallen een verkeerde diagnose ontvingen.
De waarschijnlijkheid om kurkuma, luteoline, cannabidiol, azathioprine, koper, glutathion, vitamine D en visolie in te nemen was hoger voor “gevallen” dan voor “controles”. Het is mogelijk dat er een meldings- of selectiebias bestond voor cannabidiol bij “controles”, aangezien misbruik van medicatie deelnemers kan hebben uitgesloten van ten minste enkele van de proeven die zijn opgenomen in PRO-ACT. Associaties bewijzen geen oorzakelijk verband. De acht therapieën die hier worden geïdentificeerd, zijn echter bijzonder interessant omdat er plausibele mechanismen zijn waardoor ze ALS kunnen beïnvloeden. Luteoline, glutathion en vitamine D verzwakken bijvoorbeeld oxidatieve stress en visolie, cannabidiol en azathioprine verminderen ontsteking. Bovendien was elke hier geïdentificeerde therapie tijdelijk geassocieerd met ten minste twee omkeringen. Deze therapieën zouden verder moeten worden geëvalueerd in prospectieve studies.
Ten slotte is het mogelijk dat onze gevallen genetische verschillen vertonen die ziekteresistentie verlenen. Hiervan is een precedent in HIV-elite-controllers, en die ontdekking leidde tot een effectieve behandeling, maraviroc, voor patiënten met HIV. Een mogelijkheid is dat sommige van deze patiënten mutaties hebben die leiden tot verbeterde reïnnervatie. Dit kan een bijzonder voordeel zijn bij zuiver lager motorneuronziekte, die bij onze gevallen vaak voorkwam. We zijn van plan sequenties van het gehele genoom uit te voeren op gevallen van ALS-omkeringen om te bepalen of ze genetisch verschillen van patiënten met een meer typische ALS-progressie. Door de ALS-omkeringen beter te begrijpen, hopen we ze ook vaker te kunnen laten gebeuren.
Dankwoord
De auteurs zijn alle patiënten en families van patiënten met ALS dankbaar om hun verhalen te delen in de hoop andere pALS te kunnen helpen. Zonder hun tijd en interesse was deze studie niet mogelijk geweest. We waarderen ook de hulp van Edwin Iversen, PhD en het Statistical Consulting Centre van de Duke University voor hun ondersteuning wat betreft statistische methoden. Dit werk werd ondersteund door de LVH ALS Foundation [grant nummer 1].
Vertaling: ALS Liga: Walter
Bron: Taylor & Francis