

De nouveaux médicaments pour la SLA
08-03-2018
Le diagnostic de SLA équivaut à une condamnation à mort. La maladie se manifeste de nombreuses façons. Ce qui facilite et complique à la fois le développement de médicaments. Des chercheurs mettent leur espoir dans un cocktail de médicaments nouveaux et anciens.
Par Ulrike Gebhardt
Le journaliste américain Mitch Albom rend visite tous les mardis à son ancien professeur Morrie Schwartz. Ils parlent des choses essentielles de la vie : l’amour, la vieillesse, la peur de la mort. Morrie ne vivra plus longtemps. Il souffre de la SLA.
Dans son livre ‘Mes mardis chez Morrie’, Albom décrit la maladie en sans retenue. « La SLA est comme une bougie allumée : les nerfs fondent et il reste le corps comme un tas de cire fondue. La maladie commence souvent par les jambes et s’étend vers le haut du corps. Vous perdez le contrôle des muscles des jambes, vous ne pouvez plus tenir debout. Vous perdez le contrôle des muscles de votre torse, vous ne pouvez plus vous tenir droit en position assise. Et, finalement, vous respirez à travers un tube passé à travers d’un trou dans votre cou, tandis que votre âme – entièrement consciente-reste enfermée dans cette coquille impuissante. »
La Sclérose Latérale Amyotrophique (SLA) est un des pires troubles neurologiques. « Quand vous rencontrez quelqu'un souffrant de la SLA, vous n’oubliez jamais cette expérience », dit Thomas Meyer de la Policlinique SLA de l’hôpital de la Charité à Berlin. Les symptômes sévissent d’une manière cruelle, odieuse et impitoyable.
Pourtant, le neurologue américain Jeremy Shefner (Institut neurologique Barrow Phoenix, USA) est convaincu qu’il y a des raisons d’espérer. « La recherche SLA est actuellement dans une phase passionnante et pleine d’espoir : nous sommes plus proches que jamais de nouveaux médicaments. »
Le neurologue français Jean-Martin Charcot (1825-1893) a décrit en premier la maladie en 1869. Comme directeur médical de l’hôpital de la Salpêtrière à Paris, il a traité de nombreux patients avec un large éventail de symptômes. Sigmund Freud (1856-1939), qui est resté plusieurs mois à l’hôpital du Dr Charcot, était plein d’éloges pour son « talent à observer ». Le Dr Charcot observait très minutieusement. Il ne découvrit pas seulement la SLA, mais produisit également une des premières descriptions détaillées de la sclérose en plaques.
La SLA tue les motoneurones dans le cerveau et la moelle épinière. Ces neurones, également appelés neurones efférents, commandent les muscles (voir l’illustration ci-dessous). Lorsque les muscles ne reçoivent plus d’impulsions des cellules nerveuses, ils s’affaiblissent. Certains patients sentent d’abord leurs jambes ou leurs bras se raidir ou se paralyser. D’autres malades éprouvent d’abord des difficultés à parler, à avaler ou à mastiquer. « En fin de compte, les patients SLA ne peuvent plus parler, ni faire des gestes et ni même à communiquer avec les autres par des expressions faciales », dit le neurologue Albert Ludolph de l’Hôpital Universitaire d’Ulm en Allemagne.
La maladie ne détériore pas seulement la capacité à se déplacer. Le collègue de Charcot, Pierre Marie (1853-1940) avait également observé la détérioration de certaines capacités mentales chez certains patients. « La SLA détériore le fonctionnement du lobe frontal», explique Ludolph. Elle peut provoquer ainsi des problèmes de mémoire et des changements de personnalité.
Lorsque la maladie progresse, le symptôme probablement le plus terrible apparait, l’affaiblissement des muscles respiratoires. La respiration devienne plus difficile. Plusieurs patients décèdent d’une pneumonie, ou tout simplement parce que la respiration s’arrête. On ne sait pas prédire la vitesse de progression de la maladie ni combien de temps un patient restera encore en vie après le diagnostic. La moyenne est de trois à cinq ans, mais aussi parfois plus de dix ans. L’astrophysicien britannique Stephen Hawking a vécu pendant plus de cinquante ans avec la maladie.
« La recherche est dans une phase passionnante et pleine d’espoir. Nous sommes plus proches que jamais de nouveaux médicaments »
|
RESUME MUSCLES QUI DÉPÉRISSENT 1. La SLA (sclérose latérale amyotrophique) tue les motoneurones. Les muscles dépérissent de plus en plus, provoquant la paralysie. 2. Certains défauts génétiques peuvent entraîner cette mort des cellules nerveuses, mais aussi le contact avec certaines substances chimiques. La SLA est en fait un terme générique pour diverses affections qui résultent de l’interaction entre diverses facteurs génétiques et environnementaux. 3. La SLA est considérée, jusqu'à présent, comme incurable. Mais de nouveaux médicaments sont prometteurs. Les experts attendent beaucoup des cocktails de médicaments. |
Une série d’affections
« LA SLA n’existe pas », souligne Meyer, neurologue de Berlin. « SLA est en fait un terme générique pour tout un groupe de maladies, chacune avec sa propre évolution, ses propres origines et causes. » C’est pourquoi Ammar Al-Chalabi (Kings College London) et Orla Hardiman (Trinity College Dublin) parlent plutôt d’un syndrome qui comprend plusieurs affections que d’une maladie. Les diverses manifestations de la SLA compliquent la détermination du bon diagnostic et du bon traitement. En même temps, une meilleure connaissance des différentes formes sous lesquelles se manifeste la SLA rend probablement possible de développer des traitements personnalisés qui fonctionnent mieux que les tentatives actuelles de trouver un remède pour la SLA.
Lorsque les premiers symptômes apparaissent, la moitié des cellules nerveuses sont déjà mortes
Dans cinq à dix pour cent des cas, la SLA est due à une cause génétique (voir « Les gènes défectueux »). Le gène SOD1 joue souvent un rôle, il est codant pour l’enzyme superoxyde-dismutase. Celui-ci agit comme un outil moléculaire qui neutralise les molécules d’oxygène agressives dans les cellules du corps. S’il est altéré par une mutation, il ne fonctionne plus parfaitement. Les molécules d’oxygène ont ainsi le champ libre pour attaquer. Et les cellules nerveuses y sont particulièrement sensibles. Par des changements dans leur structure, les molécules SOD peuvent également s’agglutiner ensemble, les cellules se bouchent à l’intérieur et entravent le fonctionnement des neurones. Les connaissances sur les gènes qui jouent un rôle dans la SLA conduisent à de nouveaux traitements expérimentaux. Les oligonucléotides antisens semblent bien prometteurs. Ces acides nucléiques interceptent les molécules messagères des gènes mutants, la cellule ne produisant ainsi plus de protéines nuisibles ou inutiles. Les premiers essais cliniques chez des patients SLA avec une telle mutation dans le gène SOD1 ont démontré que l’administration de ce nouveau produit dans le liquide céphalo-rachidien et dans la moelle épinière n’entraine en tous cas pas d’effets secondaires graves.
Il reste à établir si ce traitement apporte réellement une amélioration durable. Avec ces expériences, la recherche SLA joue un rôle pionnier, déclare, confiant, le neurologue Jeremy Shefner : « Si on réussit à utiliser les oligonucléotides antisens avec succès dans la lutte contre le gène SOD muté, cette approche novatrice pourra signifier une révolution pour la médecine générale. »
Pour savoir quelles autres variantes génétiques jouent un rôle dans la SLA, le programme international de recherche ‘MinE‘ a été mis en place. Les chercheurs veulent cartographier le génome de 15 000 patients SLA et de 7 500 sujets en bonne santé. Ces données précieuses sont stockées dans le superordinateur SURFsara à Amsterdam et sont rendues accessibles aux chercheurs du monde entier.
"Si vous avez une mutation dans un gène SLA, cela ne signifie pas que vous subirez nécessairement la maladie", souligne Jan Veldink de l’Université d’Utrecht, un des coordinateurs du projet MinE. « Les gènes ne sont, et de loin, pas l’explication pour tout. » Neuf cas sur dix de SLA ne peuvent être réduits à une mutation spécifique. Ils émergent de façon « sporadique ». Vous contractez la SLA si vous avez une certaine prédisposition génétique et si vous êtes exposé à certains facteurs environnementaux. « Comme le cancer, qui est une interaction entre des facteurs génétiques et toutes sortes de facteurs environnementaux » explique Thomas Meyer. « Une certaine mutation peut causer la SLA chez des personnes qui vivent en Allemagne, alors que des habitants de la Suède avec la même mutation ne contractent pas la maladie. »
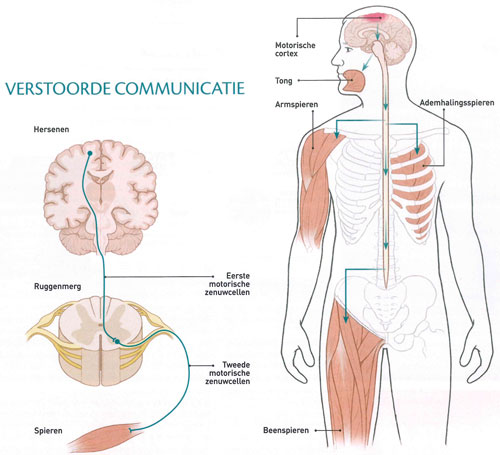
Le cerveau intiment les commandes de mouvement aux muscles par l’intermédiaire de deux groupes successifs de neurones. Le premier groupe, les motoneurones du haut, sont situés dans le cortex moteur du cerveau. Ils ont de longues extrémités, appelés axones, qui atteignent le cordon latéral (funiculus lateralis) de la moelle épinière. Ils s’y connectent avec le deuxième groupe, les motoneurones du bas. Ceux-ci se connectent, à leur tour, par de longs axones aux muscles squelettiques. Dans le cas de la SLA, les deux types de motoneurones peuvent être endommagés. Les fibres nerveuses se durcissement (en grec, ‘scleros’ signifie « dures »).
Les muscles reçoivent trop peu de signaux, ils se relâchent et se rétractent (ils ‘s’atrophient’ en jargon médical).
Les causes de la mort des cellules nerveuses ne sont toujours pas entièrement connues. On a souvent trouvé, dans les cellules malades, des amas de protéines mais ils pouvaient déjà s’y accumuler plusieurs années ou décennies avant que les premiers symptômes ne se manifestent.
Enquête dans une île tropicale
C’est pourquoi les scientifiques recherchent fébrilement à identifier des facteurs environnementaux. « Les influences environnementales sont les seuls facteurs de risque sur lesquels nous pouvons agir, même pour les familles avec une cause génétique démontrable », écrivent les Prof Ammar Al-Chalabi et Orla Hardiman. Si vous identifiez ces facteurs, vous pouvez peut-être éviter la SLA.
Une première piste vers un facteur externe a été découverte à la fin de la deuxième guerre mondiale, loin des grands centres de recherche. Les médecins américains avaient remarqué que les Chamorros, habitants de l’île de Guam dans le Pacifique Ouest, souffraient d’un taux supérieur à la moyenne de maladies neurodégénératives comme la maladie de parkinson, la démence et la SLA. Les chercheurs suspectaient l’acide aminé BMAA (bêta-N-methylamino-L-alanine) d’en être la cause. Elle est produite en des concentrations élevées par les algues bleues – vertes qui font partie de la nourriture des Chamorros. Si cette substance joue en effet un rôle, et dans l’affirmative, de quelle façon, est toujours l’objet de débats.
Les chercheurs font face à un dilemme dans leurs quêtes sur les causes. Avec le recul, il n’est souvent plus possible de mesurer quand et dans quelle doses les toxines ont agi sur un être humain. Si le génome de chacun de nous reste inchangé, notre exposome (le total de l’ensemble des interaction des facteurs environnementaux sur un être humain) est en constante évolution. Même si la science ne parvient pas à trouver un élément de preuve probante pour un produit suspect, cela ne signifie pas pour autant que la substance en question est réellement innocentée.
La liste des produits chimiques suspects dans l’environnement est longue. Les PCB (biphényles polychlorés) s’accumulent dans les tissus cérébraux, perturbent l’équilibre entre les différentes substances des signaux et font en sorte que les cellules nerveuses meurent plus vite. Les PCB ont été utilisés, dans les années 1980, dans les plastiques, les peintures et les ignifuges. Les hydrocarbures aromatiques polycycliques (HAP) sont également préjudiciables aux cellules nerveuses. Notre corps en absorbe, par exemple, en mangeant de la viande brûlée. Et certains pesticides provenant de l’agriculture peuvent perturber la communication subtilement équilibrée entre neurones, en affectant les molécules de signalisation.
Le Prof Eva Feldman, de l’Université du Michigan aux USA, a demandé à 66 patients SLA et au même nombre de sujets sains si elles avaient été en contact avec ces substances dangereuses. Et, en effet, dans le groupe SLA on a constaté un nombre au dessus de la moyenne de personnes ayant été exposés aux pesticides et engrais chimiques par leur travail ou leurs loisirs. Le Prof Feldman a publié ses résultats en 2014. Deux ans plus tard, l’équipe de recherche de Feldman a constaté des concentrations élevées de BPC, de pesticides, de pentachlorobenzène et de chlordane dans le sang des patients SLA.
Mais la prudence reste de mise. Des chercheurs italiens et grecs n’ont pu découvrir, en 2017, aucun lien clair entre la SLA et la présence de pesticides, PCB et HAP dans le liquide céphalorachidien. Ces résultats contradictoires sont typiques pour ce genre de recherche.
« À ce jour, aucun facteur environnemental n’a été clairement identifié », est la conclusion avec laquelle les Prof Al-Chalabi et Hardiman résument l’état actuel de la recherche sur les causes de la SLA. Cela ne signifie pas qu’ils ont jetté l’éponge. Ils demandent instamment des études approfondies avec un grand nombre de sujets, sur de longues périodes. C’est cher, mais c’est nécessaire. Selon ces deux chercheurs en neurosciences, de nombreuses études plus anciennes ne sont pas utilisables à cause d’erreurs méthodologiques et de points faibles.
Cocktail
D’autres scientifiques cherchent à parvenir à identifier la maladie dès les premiers stades. Car lorsque les premiers symptômes cliniques se manifestent, il peut déjà être trop tard, un tiers à la moitié des motoneurones étant déjà affectés ou morts à ce stade. Aujourd'hui, les médecins diagnostiquent la SLA au moyen d’examens neurologiques et des tests électrophysiologiques. Ceux-ci constatent la mort éventuelle de cellules nerveuses.
Il est possible qu’à l’avenir cela se fera avec des biomarqueurs dans le sang et dans le liquide céphalo-rachidien de la moelle épinière, même avant l’apparition des premiers symptômes. Plus tôt le traitement commence, plus efficace seront les médicaments.
Ces médicaments n’existent par ailleurs pas encore. La SLA reste, momentanément, incurable. Une seule panacée contre toutes les formes de SLA ne sera jamais découverte, suppose le Prof Thomas Meyer.
Vu le très grand nombre de facteurs génétiques et environnementaux, nous pouvons mieux placer nos espoirs dans un cocktail de substances différentes, qui pourrait ralentir la progression de la maladie, ou peut-être même y mettre un terme. Une telle combinaison de thérapies doit être composé de médicaments renforçant les muscles et d’autres substances prévenant la mort des cellules nerveuses. Meyer déclare : « Nous recherchons des médicaments qui, séparément, n’ont peut-être que peu d’effets, mais pourraient avoir un effet important s’ils étaient administrés ensemble». Les chercheurs travaillent sur des médicaments SLA qui devraient intervenir sur diverses facettes du processus de la maladie. La SLA est parfois décrite comme une maladie protéinique, parce que certaines protéines s’agglutiner ensemble dans les cellules nerveuses, obstruant l’intérieur des cellules. Mais, de nombreux autres processus contribuent également à la disparition des motoneurones, comme l’activation et la sécrétion de la signalisation des molécules du système immunitaire, le stress oxydatif causé par des radicaux d’oxygène libres ou un excès du neurotransmetteur glutamate avec son un effet toxique. Différents médicaments agissent sur ces facteurs. Comme l’Edaravone, un antioxydant qui réduit la quantité de radicaux d’oxygène. Ou comme le Levosimendan et le Tirasemtiv, qui renforcent les fonctions musculaires restantes. Et le NP001 qui influence l’activité des cellules immunitaires dans le cerveau. Les cellules souches seraient elles en mesure de remplacer la perte de cellules nerveuses. Celai semble une idée prometteuse, mais il reste encore beaucoup de problèmes à résoudre. Par exemple, comment amener les cellules au lieu de destination préconisé et comment les encourager à établir des connexions significatives avec d’autres cellules. Les applications réelles semblent seulement possibles dans un avenir encore lointain.
Morrie Schwartz est mort un dimanche matin en novembre 1995. À cette époque, le Riluzole était le seul médicament SLA, autorisé uniquement aux Etats-Unis. Il n’a pas pu prolonger la vie de Schwartz. Mais, en tant qu’ingrédient d’un cocktail de médicament, il aidera peut-être de nombreux autres patients dans le futur. P & B
|
LES GÈNES DÉFECTUEUX Chaque année, trois personnes sur cent mille sont diagnostiquées SLA. Dans 90 à 95 % des cas, la maladie est sous sa forme« sporadique ». Cela veut dire que la SLA est rare dans la famille du patient. Dans le reste de 5 à 10 % des cas, la maladie a une cause génétique. Dans plus de deux-tiers de cette formes génétiquement de la SLA, les chercheurs ont trouvé des mutations dans 17 gènes. Deux de ces gènes sont responsables de plus de la moitié des cas. Déjà en 1993, une mutation a été découverte dans le gène SOD1. Elle est surtout présente en Asie. Le SOD1 code la superoxyde dismutase, une enzyme qui neutralise les ions d’oxygène. Depuis 2011, nous connaissons également la mutation du gène C9orf72 (Gène sur Chromosome 9 open reading frame 72), principalement présente chez les européens. Le gène TARDBP a souvent également subi une mutation, il est codant pour la protéine dite TAR-DNA-binding 43 (TDP-43) et contribue à la lecture de l’ADN. Un autre gène qui joue un rôle dans ce domaine, FUS (‘Fused in Sarcoma’). Tout cela ne signifie pas que tous ceux qui ont une de ces mutations génétiques, subiront nécessairement la SLA. |
L’AUTEUR
ULRIKE GEBHARDT est docteure en Biologie et travaille comme journaliste scientifique à Brèmes.
PLUS A CE SUJET
The Epidemiology of ALS: A conspiracy of genes, environment and time.
Ammar AI-Chalabi et Orba Hardiman, Nature Reviews Neurology, 2013.
Association of Environmental Toxins with Amyotrophic Lateral Sclerosis.
F.-C. Su e.a. in JAMA Neurology, 2016.
Environmental Risk Factors and Amyotrophic Lateral Sclerosis (ALS): A case-control studyof ALS in Michigan.Yu Yu e.a. in PloS One, 2014.
Mes mardis avec Morrie. Mitch Albom, Edition Ambo/Anthos, 2014.
Traduction : Fabien
Source : Eos Wetenschap