

Nieuwe medicijnen voor ALS
08-03-2018
De diagnose ALS staat gelijk aan een doodvonnis. De ziekte kent vele verschijningsvormen. Dat maakt het moeilijker én makkelijker om medicijnen te ontwikkelen. Onderzoekers vestigen hun hoop op een cocktail van nieuwe en oude medicijnen.
Door Ulrike Gebhardt
Elke dinsdag gaat de Amerikaanse journalist Mitch Albom op bezoek bij zijn voormalige hoogleraar Morrie Schwartz. Ze praten over de wezenlijke dingen des levens: over de liefde, ouder worden, de angst voor de dood. Morrie zal niet lang meer leven. Hij heeft ALS.
In zijn boek Mijn dinsdagen met Morrie beschrijft Albom de ziekte op indringende wijze. 'ALS is als een brandende kaars: de zenuwen smelten weg en het lichaam blijft achter als een hoopje gesmolten was. De ziekte begint vaak bij de benen en breidt zich dan uit naar boven. Je verliest de controle over de beenspieren, zodat je niet meer kunt staan. Je verliest de controle over de spieren in je romp, zodat je niet meer rechtop kunt zitten. En uiteindelijk adem je via een buisje door een gat in je hals, terwijl je ziel – klaarwakker - opgesloten zit in een krachteloos omhulsel.'
Amyotrofische Laterale Sclerose (ALS) geldt als een van de ergste neurologische aandoeningen. 'Als je eenmaal iemand met ALS hebt ontmoet, blijft die ervaring je voor altijd bij', zegt Thomas Meyer van de ALS-polikliniek van het Charitéziekenhuis in Berlijn. De symptomen slaan op een gruwelijke en meedogenloos wrede manier toe.
Toch is de Amerikaanse neuroloog Jeremy Shefner (Barrow Neurological Institute Phoenix, VS) ervan overtuigd dat er hoop gloort aan de horizon. 'Het ALS-onderzoek bevindt zich momenteel in een spannende en hoopgevende fase: we zijn dichter bij nieuwe medicijnen dan ooit tevoren.'
De Franse neuroloog Jean-Martin Charcot (1825-1893) beschreef de ziekte in 1869 als eerste. Als geneesheer-directeur van het Hopital de la Salpêtrière in Parijs behandelde hij tal van patiënten met de meest uiteenlopende symptomen. Sigmund Freud (1856-1939), die enkele maanden in het ziekenhuis van Charcot verbleef, was vol lof over diens 'talent om te kijken'. Charcot observeerde heel nauwkeurig. Zo ontdekte hij niet alleen ALS, maar pende hij ook een van de eerste uitgebreide beschrijvingen neer van multiple sclerose.
Bij ALS sterven steeds meer motorische zenuwcellen af in de hersenen en het ruggenmerg. Deze neuronen, ook wel efferente zenuwcellen genoemd, sturen de spieren aan (zie illustratie op pag. 43). Als de spieren geen impulsen van de zenuwcellen krijgen, verzwakken ze. Sommige patiënten voelen eerst hun benen of armen verstijven of verlammen. Bij anderen begint de ziekte met spreek-, slik- of kauwproblemen. 'Uiteindelijk kunnen ALS-patiënten niet meer praten, geen gebaren meer maken en zelfs niet meer via gezichtsuitdrukkingen met anderen communiceren', zegt neuroloog Albert Ludolph van het Academisch Ziekenhuis van Ulm in Duitsland.
Niet alleen het vermogen om te bewegen wordt aangetast. Charcots collega Pierre Marie (1853-1940) had bij sommige patiënten al waargenomen dat ook bepaalde mentale vermogens achteruitgaan. 'Bij ALS functioneert de frontale kwab niet meer goed', legt Ludolph uit. Daardoor duiken geheugenproblemen op en kan je persoonlijkheid veranderen.
Naarmate de ziekte voortschrijdt, komt misschien wel het akeligste symptoom tevoorschijn. De ademhalingsspieren verzwakken. Ademen wordt steeds moeilijker. Veel patiënten overlijden door een longontsteking of doordat de ademhaling simpelweg stopt. Hoe snel dat gebeurt en hoe lang een patiënt nog leeft nadat de diagnose is gesteld, valt niet te voorspellen. Gemiddeld is dat drie à vijfjaar, maar soms ook meer dan tien jaar. De Britse astrofysicus Stephen Hawking leeft al ruim vijftigjaar met de ziekte.
'Het onderzoek bevindt zich in een spannende en hoopgevende fase. We zijn dichter bij nieuwe medicijnen dan ooit'
|
SAMENGEVAT WEGKWIJNENDE SPIEREN 1. Bij ALS (Amyotrofische Laterale Sclerose) sterven de motorische zenuwcellen af. Daardoor kwijnen ook de spieren weg, zodat je steeds meer verlamd raakt. 2. Genetische foutjes kunnen dat afsterven van de zenuwcellen veroorzaken, net als contact met bepaalde chemische stoffen. ALS is eigenlijk een verzamelnaam voor verschillende aandoeningen die het gevolg zijn van een samenspel tussen allerlei genetische en omgevingsfactoren. 3. Tot nog toe is ALS ongeneeslijk. Nieuwe middelen bieden hoop. Experts verwachten vooral veel van medicijnencocktails. |
Een verzameling van aandoeningen
'Dé ALS bestaat niet', benadrukt de Berlijnse neuroloog Meyer. 'ALS is eigenlijk een verzamelnaam voor een hele groep aandoeningen die elk hun eigen ziekteverloop, ontstaansprocessen en oorzaken hebben.' Daarom spreken Ammar Al-Chalabi (King's College Londen) en Orla Hardiman (Trinity College Dublin) liever niet van een ziekte, maar van een syndroom dat verschillende aandoeningen omvat. De uiteenlopende verschijningsvormen van ALS maken het moeilijker om de juiste diagnose te stellen en een goede behandelmethode te vinden. Tegelijk maakt meer kennis over de verschillende vormen waarin ALS zich manifesteert het wellicht mogelijk behandelingen op maat te ontwikkelen die beter werken dan de huidige pogingen om hét medicijn tegen ALS te vinden.
Als de eerste symptomen opduiken is tot de helft van de zenuwcellen al afgestorven
In vijf à tien procent van de gevallen is ALS te herleiden tot een genetische oorzaak (zie 'De foute genen' op pag. 45). Daarbij speelt vaak het gen SOD1 een rol, dat codeert voor het enzym superoxide-dismutase. Dat fungeert als een moleculair instrument dat agressieve zuurstofmoleculen in de lichaamscellen onschadelijk maakt. Als het is veranderd door een mutatie, werkt het niet meer foutloos. De zuurstofmoleculen krijgen daardoor de kans om de aanval te openen. En daar zijn vooral zenuwcellen erg gevoelig voor. Door veranderingen in hun structuur kunnen SOD-moleculen ook gaan samenklonteren, zodat de cellen van binnen verstopt raken en als nutteloos afval de werking van de neuronen belemmeren. De kennis over de genen die een rol spelen bij ALS leidt tot nieuwe experimentele therapieën. Zo lijken de zogeheten antisense-oligonucleotiden veelbelovend. Deze nucleïnezuren onderscheppen de boodschappermoleculen van het gemuteerde gen, zodat de cel niet langer schadelijke of nutteloze eiwitten kan produceren. De eerste klinische studies bij ALS-patiënten met zo'n mutatie in het SOD1-gen hebben aangetoond dat de toediening van het nieuwe medicijn in het hersenvocht in het ruggenmerg in elk geval geen ernstige bijwerkingen met zich meebrengt.
Of deze behandeling ook werkelijk een blijvende verbetering brengt, moet nog blijken. Met deze experimenten speelt het ALS-onderzoek een voortrekkersrol, zegt neuroloog Jeremy Shefner vol zelfvertrouwen. 'Als het lukt antisense-oligonucleotiden met succes in te zetten in de strijd tegen gemuteerde SOD-gen, kan deze innovatieve aanpak een revolutie betekenen voor de algehele geneeskunde.'
Om te achterhalen welke andere genvarianten een rol spelen bij ALS is het internationaal onderzoeksproject MinE opgezet. De onderzoekers willen het genoom in kaart brengen van 15.000 ALS-patiënten en 7.500 gezonde proefpersonen. Die waardevolle data worden opgeslagen in de supercomputer SURFsara in Amsterdam en zijn toegankelijk voor onderzoekers overal ter wereld.
'Als je een mutatie in een ALS-gen hebt, hoeft dat niet te betekenen dat je de ziekte ook krijgt', benadrukt Jan Veldink van de Universiteit Utrecht, een van de coördinatoren van het MinEproject. 'Genen zijn bij lange na niet de verklaring voor alles.' Negen op de tien gevallen van ALS kunnen niet herleid worden tot een specifieke mutatie. Ze doen zich 'sporadisch' voor, zoals dat heet. Je krijgt ALS als je een bepaalde genetische aanleg hebt én als je wordt blootgesteld aan bepaalde omgevingsfactoren. 'Net als bij kanker is hier sprake van een samenspel tussen genetische factoren en allerlei omgevingsfactoren', legt Thomas Meyer uit. 'Een bepaalde mutatie kan bij mensen die in Duitsland wonen ALS veroorzaken, terwijl inwoners van Zweden met dezelfde mutatie de ziekte niet krijgen.'
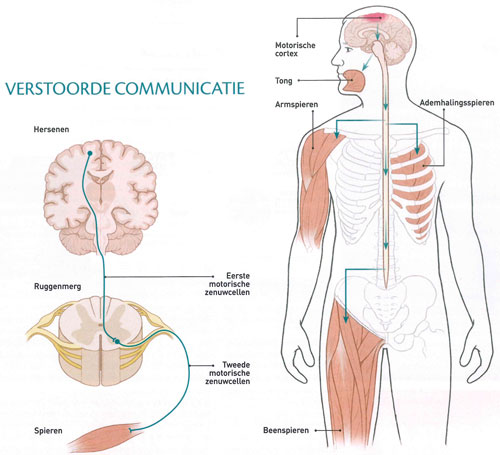 De hersenen sturen via twee achter elkaar geschakelde groepen neuronen bewegingscommando's naar de spieren. De eerste (bovenste) motorische zenuwcellen zitten in de motorische cortex van de grote hersenen. Ze hebben lange uitlopers, axonen genaamd, die tot in de funiculus lateralis (zijstreng) van het ruggenmerg reiken en zo in verbinding staan met de tweede (onderste) motorische zenuwcellen. Die maken op hun beurt via lange uitsteeksels contact met de skeletspieren.
De hersenen sturen via twee achter elkaar geschakelde groepen neuronen bewegingscommando's naar de spieren. De eerste (bovenste) motorische zenuwcellen zitten in de motorische cortex van de grote hersenen. Ze hebben lange uitlopers, axonen genaamd, die tot in de funiculus lateralis (zijstreng) van het ruggenmerg reiken en zo in verbinding staan met de tweede (onderste) motorische zenuwcellen. Die maken op hun beurt via lange uitsteeksels contact met de skeletspieren.
In geval van Amyotrofische Laterale Sclerose (ALS), een progressieve neurodegeneratieve ziekte, kunnen beide types motorische neuronen beschadigd zijn. De zenuwvezels verharden (het Griekse scleros betekent 'hard').
Daardoor ontvangen de spieren te weinig signalen, zodat ze verslappen en verschrompelen ('atrofiëren' in medisch jargon).
De oorzaken van de teloorgang van de zenuwcellen zijn nog niet geheel bekend. Vaak worden er in de zieke cellen eiwitklonters aangetroffen die zich daar misschien al jaren of decennia hebben opgehoopt voordat de eerste symptomen zich manifesteren.
Speurtocht op een tropisch eiland
Daarom zoeken wetenschappers naarstig naar die omgevingsfactoren. 'Omgevingsinvloeden zijn de enige risicofactoren die we kunnen veranderen, zelfs bij families waar een aanwijsbare genetische oorzaak aanwezig is', schrijven Ammar Al-Chalabi en Orla Hardiman. Ken je die factoren, dan kan je misschien voorkomen dat je ALS krijgt.
Een eerste spoor naar zo'n externe factor werd aan het eind van WO II ontdekt -ver verwijderd van de grote onderzoekscentra. Amerikaanse artsen hadden opgemerkt dat de Chamorro, de bewoners van het eiland Guam in het westen van de Stille Oceaan, bovengemiddeld vaak kampten met neurodegeneratieve ziektes als parkinson, dementie en ALS. De verdenking van de onderzoekers viel op het aminozuur BMAA (bèta-N-methylamino-L-alanine) dat wordt geproduceerd door blauwalgen en in hoge concentraties voorkwam in het voedsel van de Chamorro. Of deze stof inderdaad een rol speelt, en zo ja, op welke manier, is nog altijd voorwerp van debat.
In de zoektocht naar de oorzaken staan onderzoekers voor een dilemma. Achteraf is vaak niet meer te meten welke giftige stoffen wanneer en in welke doses op een mens hebben ingewerkt. Terwijl het genoom van u en ik vaststaat, verandert ons zogeheten exposoom voortdurend. Dat is het totaal van alle op een mens inwerkende omgevingsfactoren. Als de wetenschap dus voor een bepaalde verdachte stof geen eenduidig bewijs kan vinden, hoeft dat niet te beteken dat de stof in kwestie ook werkelijk onschuldig is.
De lijst van verdachte chemicaliën die in het milieu voorkomen, is lang. pcb's (polychloorbifenyl) hopen zich op in het hersenweefsel, verstoren het evenwicht tussen de verschillende signaalstoffen en zorgen ervoor dat zenuwcellen sneller afsterven. Pcb's zijn tot in de jaren 1980 gebruikt in onder meer kunststoffen, verf en brandvertragers. Polycyclische aromatische koolwaterstoffen (PAK's) zijn eveneens funest voor zenuwcellen. Die stoffen komen bijvoorbeeld in ons lichaam terecht via het eten van aangebrand vlees. En sommige pesticiden uit de landbouw kunnen de subtiel uitgebalanceerde communicatie tussen de neuronen verstoren doordat ze de signaalstoffen beïnvloeden.
Eva Feldman (universiteit van Michigan, VS) vroeg 66 ALSpatiënten en hetzelfde aantal gezonde proefpersonen of ze met zulke gevaarlijke stoffen in aanraking waren geweest. En inderdaad, in de ALS-groep bleken bovengemiddeld veel proefpersonen in hun werk of hun vrije tijd te zijn blootgesteld aan chemische meststoffen en gewasbeschermingsmiddelen. Feldman publiceerde haar resultaten in 2014. Twee jaar later kon Feldmans onderzoeksteam in het bloed van ALS-patiënten verhoogde concentraties pcb's aantonen, alsook de pesticiden pentachloorbenzeen en chlordaan.
Toch is voorzichtigheid op zijn plaats. Italiaanse en Griekse onderzoekers konden in 2017 geen duidelijke samenhang ontdekken tussen ALS en de aanwezigheid van pesticiden, pcb's en PAK's in het ruggenmergvocht Zulke tegenstrijdige resultaten zijn kenmerkend voor dit soort onderzoeken.
'Tot op heden is er geen eenduidige omgevingsfactor gevonden', luidt de conclusie waarmee Al-Chalabi en Hardiman de huidige stand van het onderzoek naar de oorzaken van ALS samenvatten. Dat betekent niet dat ze de handdoek in de ring gooien. Ze dringen juist aan op omvangrijke studies met grote aantallen proefpersonen, over langere periodes. Dat is duur, maar noodzakelijk. Veel oudere studies zijn niet goed bruikbaar wegens methodologische fouten en tekortkomingen, aldus de beide neurowetenschappers.
Cocktail
Andere wetenschappers zoeken naar methodes om de ziekte in een heel vroeg stadium op het spoor te komen. Want als de eerste klinische symptomen zich manifesteren, kan het al te laat zijn, aangezien dan al een derde tot de helft van de motorische neuronen is aangetast of afgestorven. Vandaag diagnosticeren artsen ALS met behulp van neurologische onderzoeken en elektrofysiologische tests. Die stellen vast of er zenuwcellen afsterven.
Mogelijk kan dat in de toekomst aan de hand van biomarkers in het bloed en in het hersenvocht in het ruggenmerg. Nog voordat de eerste symptomen optreden. Hoe vroeger de behandeling start, hoe effectiever de medicijnen.
Zulke medicijnen zijn er trouwens nog niet. ALS is nog altijd ongeneeslijk. Eén enkel wondermiddel tegen alle vormen van ALS zullen we nooit vinden, denkt Thomas Meyer.
Gezien het enorme aantal genetische en omgevingsfactoren kunnen we onze hoop beter vestigen op een cocktail van verschillende stoffen, die dan als geheel het voortschrijden van de ziekte zou kunnen vertragen of misschien zelfs een halt toeroepen. Zo'n combinatietherapie moet dan bestaan uit middelen die de spieren versterken en andere stoffen die het afsterven van zenuwcellen tegengaan. 'We zijn op zoek naar medicijnen die elk op zich misschien weinig uithalen, maar als collectief een groot effect kunnen hebben', aldus Meyer. Onderzoekers werken aan ALS-medicijnen die op verschillende plaatsen in het ziekteproces moeten ingrijpen. ALS wordt wel eens een eiwitziekte genoemd, omdat bepaalde eiwitten in de zenuwcellen samenklonteren zodat de cellen van binnenuit verstopt raken. Tal van andere processen dragen ook hun steentje bij aan de teloorgang van de motorische zenuwcellen, zoals de activering en uitscheiding van signaalstoffen van het immuunsysteem, oxidatieve stress veroorzaakt door vrije zuurstofradicalen en een overmaat van de neurotransmitter glutamaat, die daardoor een toxisch effect krijgt. Verschillende medicijnen werken in op die factoren. Zo is er de stof edaravone, een antioxidant die de hoeveelheid zuurstofradicalen vermindert. Levosimendan en Tirasemtiv hebben de taak de nog resterende spierfunctie te versterken. En NP001 beïnvloedt de activiteit van immuuncellen in het brein. Stamcellen zouden verloren gegane zenuwcellen kunnen vervangen. Dat lijkt een veelbelovend idee, maar er moeten nog veel problemen worden opgelost. Bijvoorbeeld de vraag hoe de cellen op de plaats van bestemming moeten raken en hoe we hen er dan toe kunnen aanzetten zinvolle verbindingen met andere cellen te vormen. Echte toepassingen lijken nog verre toekomstmuziek.
Morrie Schwartz overleed op een zondagochtend in november 1995. Op dat moment was het enige ALS-medicijn, riluzole, pas toegelaten in de Verenigde Staten. Het heeft Schwartz' leven niet meer kunnen verlengen. Maar als ingrediënt van een medicijnencocktail zal het in de toekomst misschien veel andere patiënten helpen. P&B
|
DE FOUTE GENEN Jaarlijks krijgen drie op de honderdduizend mensen de diagnose ALS. In 90 tot 95 procent van de gevallen is de ziekte 'sporadisch'. Dat wil zeggen dat ALS in de familie van de patiënt niet vaak voorkomt. In de overige 5 tot 10 procent van de gevallen heeft de ziekte een genetische oorzaak. Bij ruim twee derde van deze genetisch bepaalde vormen van ALS hebben onderzoekers mutaties in 17 genen aangetroffen. Twee van die genen zijn verantwoordelijk voor meer dan de helft van de gevallen. Al in 1993 is een mutatie ontdekt in het gen SOD1. Ze komt vooral in Azië voor. SOD1 codeert voor superoxide-dismutase, een enzym dat zuurstofionen onschadelijk maakt. Sinds 2011 kennen we de mutatie in het gen C9orf72 (chromosome 9 open reading frame 72). Die komt vooral bij Europeanen voor. Vaak is ook het gen TARDBP gemuteerd, dat codeert voor het zogeheten TAR-DNA-bindende eiwit 43 (afgekort TDP-43) en een bijdrage levert aan het aflezen van het DNA. Een ander gen dat daarbij een rol speelt, is FUS (een afkorting van fused in sarcoma). Dat alles betekent niet dat iedereen die een mutatie in deze genen heeft, noodzakelijkerwijs ALS krijgt. |
DE AUTEUR
ULRIKE GEBHARDT is doctor in de biologie en werkt als wetenschapsjournalist in Bremen.
MEER OVER DIT ONDERWERP
The Epidemiology of ALS: A conspiracy of genes, environment and time.
Ammar AI-Chalabi en Orba Hardiman in Nature Reviews Neurology, 2013.
Association of Environmental Toxins with Amyotrophic Lateral Sclerosis.
F.-C. Su e.a. in JAMA Neurology, 2016.
Environmental Risk Factors and Amyotrophic Lateral Sclerosis (ALS): A case-control studyof ALS in Michigan. Yu Yu e.a. in PloS One, 2014.
Mijn dinsdagen met Morrie. Mitch Albom, Uitgeverij Ambo/Anthos, 2014.
Bron: Eos Wetenschap